Press release

NOBELFÖRSAMLINGEN KAROLINSKA INSTITUTET
THE NOBEL ASSEMBLY AT THE KAROLINSKA INSTITUTE
6 October 1997
The Nobel Assembly at the Karolinska Institute has today decided to award the Nobel Prize in Physiology or Medicine for 1997 to
Stanley B. Prusiner
for his discovery of “Prions – a new biological principle of infection”.
Summary
The 1997 Nobel Prize in Physiology or Medicine is awarded to the American Stanley Prusiner for his pioneering discovery of an entirely new genre of disease-causing agents and the elucidation of the underlying principles of their mode of action. Stanley Prusiner has added prions to the list of well known infectious agents including bacteria, viruses, fungi and parasites. Prions exist normally as innocuous cellular proteins, however, prions possess an innate capacity to convert their structures into highly stabile conformations that ultimately result in the formation of harmful particles, the causative agents of several deadly brain diseases of the dementia type in humans and animals. Prion diseases may be inherited, laterally transmitted, or occur spontaneously. Regions within diseased brains have a characteristic porous and spongy appearance, evidence of extensive nerve cell death, and affected individuals exhibit neurological symptoms including impaired muscle control, loss of mental acuity, memory loss and insomnia. Stanley Prusiner’s discovery provides important insights that may furnish the basis to understand the biological mechanisms underlying other types of dementia-related diseases, for example Alzheimer’s disease, and establishes a foundation for drug development and new types of medical treatment strategies.
In 1972 Stanley Prusiner began his work after one of his patients died of dementia resulting from Creutzfeldt-Jakob disease (CJD). It had previously been shown that CJD, kuru, and scrapie, a similar disease affecting sheep, could be transmitted through extracts of diseased brains. There were many theories regarding the nature of the infectious agent, including one that postulated that the infectious agent lacked nucleic acid, a sensational hypothesis since at the time all known infectious agents contained the hereditary material DNA or RNA. Prusiner took up the challenge to precisely identify the infectious agent and ten years later in 1982 he and his colleagues successfully produced a preparation derived from diseased hamster brains that contained a single infectious agent. All experimental evidence indicated that the infectious agent was comprised of a single protein, and Prusiner named this protein a prion, an acronym derived from “proteinaceous infectious particle.” It should be noted that the scientific community greeted this discovery with great skepticism, however, an unwavering Prusiner continued the arduous task to define the precise nature of this novel infectious agent.
The infectious prion particle forms within the body
Where was the gene encoding the prion, the piece of DNA that determined the sequence of the amino acids comprising the prion protein? Perhaps the gene was closely associated with the protein itself as in a small virus? The answers to these questions came in 1984 when Prusiner and colleagues isolated a gene probe and subsequently showed that the prion gene was found in all animals tested, including man. This startling finding raised even more questions. Could prions really be the causative agent of several dementia-type brain diseases when the gene was endogenous to all mammals? Prusiner must have made a mistake! The solution to this problem became evident with the sensational discovery that the prion protein, designated PrP, could fold into two distinct conformations, one that resulted in disease (scrapie PrP = PrPSc) and the other normal (PrP = PrPc). It was subsequently shown that the disease-causing prion protein had infectious properties and could initiate a chain reaction so that normal PrPc protein is converted into the more stabile PrPSc form. The PrPSc prion protein is extremely stabile and is resistant to proteolysis, organic solvents and high temperatures (even greater than 100o C). With time, non-symptomatic incubation periods vary from months to years, the disease-causing PrPSc can accumulate to levels that result in brain tissue damage. In analogy to a well known literary work, the normal PrPc can be compared to the friendly Dr. Jekyll and the disease causing PrPSc to the dangerous Mr. Hyde, the same entity but in two different manifestations.
Mutations in the prion gene cause hereditary brain diseases
The long incubation time for prion based disease hampered the initial efforts to purify the prion protein. In order to assess purification schemes Prusiner was forced to use scores of mice and in each experiment wait patiently for approximately 200 days for the appearance of disease symptoms. The purification efforts accelerated when it was demonstrated that scrapie could be transferred to hamsters, animals that exhibited markedly shortened incubation times. Together with other scientists, Prusiner cloned the prion gene and demonstrated that the normal prion protein was an ordinary component of white blood cells (lymphocytes) and was found in many other tissues as well. Normal prion proteins are particularly abundant on the surface of nerve cells in the brain. Prusiner found that the hereditary forms of prion diseases, CJD and GSS (see the last section), were due to mutations in the prion gene. Proof that these mutations caused disease was obtained when the mutant genes were introduced into the germline of mice. These transgenic mice came down with a scrapie-like disease. In 1992 prion researchers obtained conclusive evidence for the role of the prion protein in the pathogenesis of brain disease when they managed to abolish the gene encoding the prion protein in mice, creating so called prion knock-out mice. These prion knock-out mice were found to be completely resistant to infection when exposed to disease-causing prion protein preparations. Importantly, when the prion gene was reintroduced into these knock-out mice, they once again became susceptible to infection. Strangely enough, mice lacking the prion gene are apparently healthy, suggesting that the normal prion protein is not an essential protein in mice, its role in the nervous system remains a mystery.
Structural variant disease-causing prions accumulate in different regions of the brain
Specific mutations within the prion gene give rise to structurally variant disease-causing prion proteins. These structural prion variants accumulate in different regions of the brain. Dependent upon the region of the brain that becomes infected, different symptoms, typical for the particular type of disease are evident. When the cerebellum is infected the ability to coordinate body movements declines. Memory and mental acuity are affected if the cerebral cortex is infected. Thalamus specific prions disturb sleep leading to insomnia, and prions infecting the brain stem primarily affect body movement.
Other dementias may have a similar background
Prusiner’s pioneering work has opened new avenues for understanding the pathogenesis of more common dementia-type illnesses. For example, there are indications that Alzheimer’s disease is caused when certain, non-prion, proteins undergo a conformational change that leads to the formation of harmful deposits or plaques in the brain. Prusiner’s work has also established a theoretical basis for the treatment of prion diseases. It may be possible to develop pharmacological agents that prevent the conversion of harmless normal prion proteins to the disease-causing prion conformation.
Intrinsic defense mechanisms do not exist against prions
Prions are much smaller than viruses. The immune response does not react to prions since they are present as natural proteins from birth. They are not poisonous, but rather become deleterious only by converting into a structure that enables disease causing prion proteins to interact with one another forming thread-like structures and aggregates that ultimately destroy nerve cells. The mechanistic basis underlying prion protein aggregation and their cummulative destructive mechanism is still not well understood. In contrast to other infectious agents, prion particles are proteins and lack nucleic acid. The ability to transmit a prion infection from one species to another varies considerably and is dependent upon what is known as a species barrier. This barrier reflects how structurally related the prions of different species are.
Prion diseases in animals and man
Without exception, all known prion diseases lead to the death of those affected. There are, however, great variations in pre-symptomatic incubation times and how aggressively the disease progresses.
Scrapie, a prion disease of sheep, was first documented in Iceland during the 18th century. Scrapie was transferred to Scotland in the 1940s. Similar prion diseases are known to affect other animals, e.g., mink, cats, deer and moose.
Bovine Spongiform Encephalopathy (BSE) – Mad cow disease is a prion disease that has recently received a great deal of publicity. In England BSE was transmitted to cows through feedstuff supplemented with offals from scrapie-infected sheep. The BSE epidemic first became evident in 1985. Due to the long incubation time the epidemic did not peak until 1992. In this year alone roughly 37,000 animals were affected.
Kuru among the Fore-people in New Guinea was studied by Carleton Gajdusek (recipient of the 1976 Nobel Prize in Physiology or Medicine). Kuru was shown to be transmitted in connection with certain cannibalistic rituals and was thought to be due to an unidentified “slow virus”. The infectious agent has now been identified as a prion. Duration of illness from first symptoms to death: 3 to 12 months.
Gertsmann-Sträussler-Scheinker (GSS) disease is a hereditary dementia resulting from a mutation in the gene encoding the human prion protein. Approximately 50 families with GSS mutations have been identified. Duration of illness from evidence of first symptoms to death: 2 to 6 years.
Fatal Familial Insomnia (FFI) is due to another mutation in the gene encoding the human prion protein. Nine families have been found that carry the FFI mutation. Duration of illness from evidence of first symptoms to death: roughly one year.
Creutzfeldt-Jakob Disease (CJD) affects about one in a million people. In 85-90% of the cases it has been shown that CJD occurs spontaneously. Ten to fifteen per cent of the CJD cases are caused by mutations in the prion protein gene. In rare instances CJD is the consequence of infection. Previously infections were transmitted through growth hormone preparations prepared from the pituitary gland of infected individuals, or brain membrane transplants. About 100 families are known carriers of CJD mutations. Duration of illness from evidence of first symptoms to death: roughly one year.
A new variant of CJD that may have arisen through BSE-transmission. Since 1995 about 20 patients have been identified that exhibit CJD-like symptoms. Psychological symptoms with depression have dominated, but involuntary muscle contractions and difficulties to walk are also common.
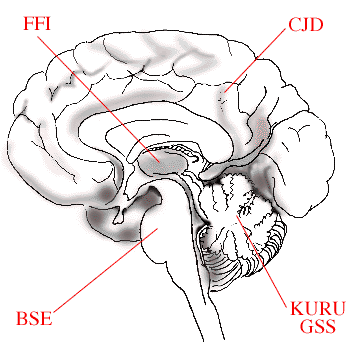
The figure schematically illustrates how variants of disease causing prions affect different parts of the brain. In Bovine Spongiform Encephalitis (BSE), the brain stem is affected, in Fatal Familial Insomnia (FFI), the thalamus region, in Creutzfeldt-Jakob Disease (CJD), the cerebral cortex, while in KURU and Gerstmann-Sträussler-Scheinker (GSS) disease the cerebellum is damaged.
Nobel Prizes and laureates
Six prizes were awarded for achievements that have conferred the greatest benefit to humankind. The 14 laureates' work and discoveries range from quantum tunnelling to promoting democratic rights.
See them all presented here.

