Advanced information
Scientific Background:
How cells sense and adapt to oxygen availability (pdf)
![]()
Scientific Background
How cells sense and adapt to oxygen availability
The Nobel Prize in Physiology or Medicine for 2019 is awarded to William Kaelin, Jr., Sir Peter Ratcliffe, and Gregg Semenza. The need for oxygen to sustain life has been understood since the onset of modern biology; but the molecular mechanisms underlying how cells adapt to variations in oxygen supply were unknown until the prize-winning work described here. Animal cells undergo fundamental shifts in gene expression when there are changes in the oxygen levels around them. These changes in gene expression alter cell metabolism, tissue re-modeling, and even organismal responses such as increases in heart rate and ventilation. In studies during the early 1990’s, Gregg Semenza identified, and then in 1995 purified and cloned, a transcription factor that regulates these oxygen-dependent responses. He named this factor HIF, for Hypoxia Inducible Factor, and showed that it consists of two components: one a novel and oxygen-sensitive moiety, HIF-1α, and a second, previously identified and constitutively expressed and non-oxygen-regulated protein known as ARNT. William Kaelin, Jr. was in 1995 engaged in the study of the von Hippel-Lindau tumor suppressor gene, and after isolation of the first full-length clone of the gene showed that it could suppress tumor growth in VHL mutant tumorigenic cell lines. Ratcliffe then demonstrated in 1999 that there was an association between VHL and HIF-1α, and that VHL regulated HIF-1α post-trans-lational and oxygen-sensitive degradation. Finally, the Kaelin and Ratcliffe groups simultaneously showed that this regulation of HIF-1α by VHL depends on hydroxylation of HIF-1α, a covalent modification that is itself dependent on oxygen. Through the combined work of these three laureates it was thus demonstrated that the response by gene expression to changes in oxygen is directly coupled to oxygen levels in the animal cell, allowing immediate cellular responses to occur to oxygenation through the action of the HIF transcription factor.
Oxygen and Animal Life
In the early 1770’s, the Swedish scientist Carl Scheele determined that, from his calculations, approximately one fourth of the volume of air was ‘feuer luft’, or ‘fire air’, as he called it; that is to say, the component of the atmosphere that allows substances to burn. This was eventually published in 1777 (Scheele, 1777). At essentially the same time in England, Joseph Priestley also found a method to purify this previously unknown gas, calling it de-phlogisticated air (Priestley, 1775). Antoine Lavoisier was simultaneously with Scheele and Priestley carrying out experiments to isolate this substance in Paris, and Lavoisier gave the gas the name we know it by today: oxygen (Lavoisier, 1777).
Oxygen is necessary for animal life during the oxidation reactions that drive the conversion of nutrients in food to ATP. Indeed, calibrating cellular conditions to the amount of available oxygen is a critical aspect of controlling meta-bolism. This has been known for more than a century; for example, in 1858 Louis Pasteur was the first to show that there is a complex balance of oxygen use in animal cells, and that cells use multiple pathways to accomplish energy con-version (Pasteur, 1858). The mechanisms under-lying oxygen sensing in animals have been previously marked by two Nobel Prizes from more than 75 years ago: to Otto Warburg in 1931 for his discoveries concerning the enzymatic basis for cellular respiration, and to Corneille Heymans in 1938 for his findings on the role of the nervous system in the respiratory response to oxygen. However, for most of the 20th century, it was not clear how adaptations to oxygen flux were regulated at the fundamental level of gene expression.
Adaptation to variations in oxygen
In almost all animal cells, the ability to rapidly respond and adapt to variations in oxygen availability is essential. It is clear from studies of molecular taxonomy that during evolution, as animal cells began organizing themselves into multicellular three-dimensional structures, this response to oxygen flux became more than a cell-autonomous reaction allowing metabolic adaptations within individual cells; it also allowed the development of complex physiological responses. Cells need to adapt in many auto- nomous ways to variations in oxygen levels, in particular by adjusting their metabolic rates. When we examine this response at the level of tissues and organs, we find that multi-cellular organisms need to both remodel tissues to adapt to altered oxygen levels (for example, by reconstructing vasculature following injury) and adapt the whole organism to compensate for changes in oxygenation (e.g., the increased ventilatory responses seen during exercise, or at exposure to high altitude).
As an example: in humans at high altitude, variations in oxygen levels in the blood are sensed by specialized cells in our kidneys that make and release the hormone erythropoietin (EPO). This hormone activates red blood cell synthesis (erythropoiesis) in the bone marrow. One way of triggering this reaction is to be exposed to the low oxygen levels of high altitude: living at high altitudes boosts EPO production by the kidney, leading to increased concentration of erythrocytes in our blood, which in turn helps us adapt to lower oxygen partial pressures.
Animals can be exposed to low oxygen environments, but importantly, oxygen levels vary in tissues as well. Tissue oxygen levels in animals vary both spatially and temporally; and this variation occurs during normal physiological events (drops in available oxygen in skeletal muscle during exertion, for example) as well as in pathological processes such as cancer and infection. It became clear from research in the 1970’s and 1980’s that these local and transient variations in oxygen partial pressure regulate critical adaptive responses in both cells and tissues through changes in gene transcription. These gene regulatory responses alter cellular metabolism, and control fundamental develop-mental, regenerative and defense processes, including those as diverse as angiogenesis, inflammation, and development.
This ability of animal cells to sense different concentrations of oxygen and, as a result, re-wire their gene expression patterns, is essential for the survival of virtually all animals. The oxygen-activated signaling pathways that are controlled by these pathways affect at least 300 genes, belonging to a wide variety of regulatory networks. These molecular pathways pervade numerous physiological processes, ranging from organ development and metabolic homeostasis to tissue regeneration and immunity, and play important roles in many diseases, including cancer.
Oxygen and the erythropoietic response
Any signaling pathway of profound importance to animal life will almost certainly turn out to involve numerous layers of fine-tuning and points of intersection with other molecular pathways. The oxygen response pathway is no exception. As expected, therefore, the molecular details of oxygen response regulation did not stop unfolding once the discoveries now awarded with the 2019 Nobel Prize had been made. On the contrary, these key discoveries opened the field, and led to an explosion of work that has uncovered an immense molecular complexity to the response to oxygen flux.
The fundamental discoveries of Gregg Semenza, William Kaelin and Sir Peter Ratcliffe all revolve around the actions of the HIF (hypoxia inducible factor) transcription factor. The discovery of this factor had its roots in the work done by a number of people in 1986 and 1987, including Maurice Bondurant, Mark Koury, and Jaime Caro; their work showed that hypoxia causes increases in the transcriptional expression of the erythropoietin hormone (EPO) in kidney (Bondurant and Koury, 1986; Jelkmann and Hellwig-Burgel, 2001; Schuster et al., 1987). This finding in turn had its roots in experiments going back to 1882 and the French physiologist Paul Bert, who first demon-strated the cardiovascular effects of hypoxia (Bert, 1878), and was the first to show that exposure to high altitude increased red blood cell counts (Bert, 1882).
The isolation of HIF
Once it was realized that the EPO gene showed a hypoxia-induced transcriptional response, the next step was to determine the actual DNA sequence in the EPO gene regulatory region that was responsible for the oxygen sensitivity. Semenza decided to trace the transcriptional regulatory elements of the EPO gene in transgenic mice, using clones of different size DNA fragments encompassing the human EPO gene. Semenza and colleagues first demonstrated that a 4 kilobase region covering the EPO-coding sequence, plus some small 5’ and 3’ flanking sequences, led to EPO production in all transgenic tissues analyzed, and caused increased circulating EPO levels and a resultant polycythemia, i.e., increased red blood cell counts (Semenza et al., 1989). He next showed that a longer EPO gene construct containing 6 kilobases of 5’ flanking DNA was able to confer inducible EPO expression in the kidney (Semenza et al., 1990). This work pointed to a complex transcriptional regulation of EPO response to oxygen, including both positive and negative regulatory elements.
A year later, in 1991, Semenza published two additional studies that added significant infor mation about the regulation of the EPO gene: 1) a DNAse hypersensitivity study pinpointed a small region in the EPO 3’ flanking DNA that bound several nuclear factors, at least two of which were induced in liver and kidney by anemia; this small region was able to function as a hypoxia-inducible enhancer in transient expression assays in vitro (Semenza et al., 1991b); 2) a study that further characterized the transcriptional regulation of EPO in transgenic models (Semenza et al., 1991a). At about the same time, work from Sir Peter Ratcliffe’s and Jaime Caro’s laboratories reported on the presence of a cis-acting DNA element 3’ of the EPO gene that conferred oxygen-responsiveness to reporter constructs transfected to cultured hepatoma cells (Beck et al., 1991; Pugh et al., 1991).
The work summarized above led Semenza in 1992 to identify an approximately 50 base pair enhancer at the 3’ end of the EPO gene that could be used to elicit hypoxia-inducible reporter gene expression in cultured cells. This enhancer, which Semenza termed a hypoxia response element (HRE), bound several nuclear factors in hepatoma cells: one that was constitutive, and one that was induced by low oxygen levels (hypoxia). The latter factor was consequently termed the hypoxia-inducible factor (HIF) by Semenza (Semenza and Wang, 1992).
Both Ratcliffe and Semenza were able to demonstrate that the EPO 3’ enhancer could drive hypoxia-induced reporter expression in a wide range of mammalian cell types (Maxwell et al., 1993; Wang and Semenza, 1993). This showed that the molecular mechanism involved in oxygen regulation of the EPO gene was active in a diverse range of animal cells, a finding that opened up the possibility that this new factor represented part of a common cellular machinery for oxygen sensing.
The notion that hypoxic HIF induction could be observed in many types of mammalian cells, and not only the EPO-producing cells in the kidney and liver, sparked the interest and attention of a wider community of scientists. The discovery of HIF indicated the potential existence of a universal molecular machinery underlying metabolic adaptation and the induction of tissue remodeling in response to tissue oxygen flux.
At this point, Semenza took a biochemical approach to purify the protein from large quantities of cell extracts. As the functional assay for HIF during its purification, he used electrophoretic mobility shift assays (EMSA) applied to the 3’ enhancer element of the EPO gene (Wang et al., 1995; Wang and Semenza, 1995). Amino acid sequencing and subsequent cDNA cloning of the purified proteins revealed that HIF itself was a heterodimer, composed of two different gene products. The first of these components was the oxygen sensitive part of the HIF factor, termed by Semenza HIF-1α; and the second component was a constitutively expressed gene that was initially termed HIF-1β, until it became evident that this second component had been previously cloned and described, as the Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT) (Wang et al., 1995). The ARNT protein heterodimerizes with a number of other factors, and since its expression was not oxygen-sensitive, it quickly became clear that HIF-1α was the regulator of oxygen responsiveness in the HIF complex.
The HIF family broadens
A protein that is highly related to HIF-1α was cloned independently by four different groups, i.e., those of Yoshiaki Fujii-Kuriyama, Werner Risau, Christopher Bradfield, and Steven McKnight, all in 1997 (Ema et al., 1997; Flamme et al., 1997; Hogenesch et al., 1997; Tian et al., 1997); it had a number of names initially, including one still commonly used (HIF-2a), but the gene is properly designated EPAS1. The EPAS1 gene encodes a protein that has a high degree of sequence homology to HIF-1α, and also binds to ARNT as a heterodimer, it shares HIF-1α’s sensitivity to hypoxia, and has essentially all of the same regulatory motifs as those described below for HIF-1α.
There are, however, significant differences in function between HIF-1α and EPAS1. The HIF-1α gene deletion in mice gives rise to a clear phenotype, i.e., mid-gestational lethality (Iyer et al., 1998; Ryan et al., 1998); but Epas1 gene deletions have highly varying phenotypes, likely due to variations in genetic background (Compernolle et al., 2002; Peng et al., 2000; Tian et al., 1998). Additionally, there is a wealth of evidence that some hypoxic responses are exclusively controlled by one or the other oxygen-sensitive HIF isoform; erythropoiesis, for example, appears primarily controlled by EPAS1 (Fandrey, 2004).
Regulation of HIF is post-translational and involves VHL
Data obtained by a number of laboratories, including Ratcliffe’s, showed that HIF-1α levels were themselves regulated by changes in protein stability, and not by changes in gene transcription or protein synthesis (Huang et al., 1998a; Kallio et al., 1999; Pugh et al., 1997; Salceda and Caro, 1997; Srinivas et al., 1999). It was further demon strated by a number of groups, including those of Caro and H. Frank Bunn, that HIF-1α was degraded through the ubiquitin-proteasome pathway, and that this occurred in an oxygen-dependent manner (Huang et al., 1998b; Salceda and Caro, 1997). This work also identified the specific structural domain in HIF-1α responsible for its oxygen-dependent degradation (termed the ODD region of the protein, and present in both HIF-1α and EPAS1).
At approximately this point, in 1995, Kaelin’s group published the first full-length sequence of the VHL tumor suppressor gene, and showed that re-introducing a wild type version of VHL into a renal carcinoma cell line prevented the cell line from forming tumors (Iliopoulos et al., 1995). Kaelin and a number of other groups had been studying the VHL gene and its link to a number of families with a genetic predisposition to certain cancers. The paper from Kaelin established that VHL is a tumor suppressor gene, whose activity can act to inhibit tumor growth of cells from patients with VHL mutations. In 1996, during the course of the characterization of the VHL gene, collaborative work between Kaelin’s group and the group of Mark Goldberg showed that a number of HIF target genes were over-expressed in VHL mutant cell lines (Iliopoulos et al., 1996). This finding suggested that the two pathways, of HIF response and VHL-linked tumorigenesis, could be linked in some fashion.
An important clue regarding VHL function next came with the identification of binding partners to the VHL protein. Richard Klausner and colleagues, as well as Kaelin and his group, had found in 1995 that VHL interacts with the transcriptional elongation factors elongins B and C (Duan et al., 1995; Kibel et al., 1995), and in 1997, Klausner, W. Marston Linehan, and colleagues showed that VHL is found in a complex with the Cul-2 protein (Pause et al., 1997), a factor involved in protein ubiquitination; a finding subsequently replicated by Kaelin (Lonergan et al., 1998). Since elongin C and Cul-2 show structural similarity to Skp1 and Cdc53, factors that were known to target specific proteins for ubiquitin-dependent proteolysis, these observations revealed a potential link between VHL and protein degradation.
HIF is targeted for ubiquitination and proteo-lysis by VHL
While during the period between 1996 and 1998 it was made clear that HIF-1α and EPAS1 get rapidly eliminated by proteasomal degradation at normoxia, it was still unknown how this process was inhibited during hypoxia. A missing piece of the puzzle was the ubiquitin E3 ligase suspected to be involved in targeting HIF-1α for degradation. Here, Ratcliffe and colleagues provided a key breakthrough in 1999, in a landmark paper where they showed that the VHL complex is involved in HIF-1α proteolysis (Maxwell et al., 1999). They and others subsequently showed that VHL acts as a recognition component of a ubiquitin E3 ligase complex in this process (Cockman et al., 2000; Kamura et al., 2000; Krieg et al., 2000; Ohh et al., 2000; Tanimoto et al., 2000).
A critical and remaining piece of the puzzle at that point was how VHL-HIF-1α interaction and subsequent HIF-1α degradation was regulated by oxygen. Importantly, the Maxwell et al., paper from 1999 points out that VHL-HIF-1α interaction requires an activity that is both oxygen and iron-dependent. This finding initiated the search for the mechanism: both for the oxygen-dependent chemical modification of HIF-1α that enables VHL binding, and the enzyme(s) that catalyze that reaction.
At that time, oxygen-dependent protein hydroxylation was known to occur in collagen proteins, and was known to be mediated by collagen prolyl-4-hydroxylase. Thus, it was suspected that oxygen-dependent hydroxylation of proline residues in HIF-1α might confer the conformational change required to allow VHL binding. This turned out to be the case. In 2001, the Ratcliffe and Kaelin laboratories simul-taneously reported that oxygen-dependent 4-hydroxylation of two proline residues within the ODD domain of HIF-1α increased the affinity for VHL-complex binding of the HIF transcription factor. The two papers describing this were published back-to-back (Ivan et al., 2001; Jaakkola et al., 2001).
The oxygen-dependent switches
Proline hydroxylation requires oxygen, and thus the elegant mechanism of post-translational regulation of HIF-1α and EPAS1 proteins was revealed: in the absence of oxygen, no hydroxylation can occur, and VHL does not recognize HIF-1α; because of this, HIF-1α is not ubiquitinated, and so avoids proteasomal degradation and remains intact. It can then accumulate, and transcriptionally activate the hypoxia-induced gene α program (see Figure 1).
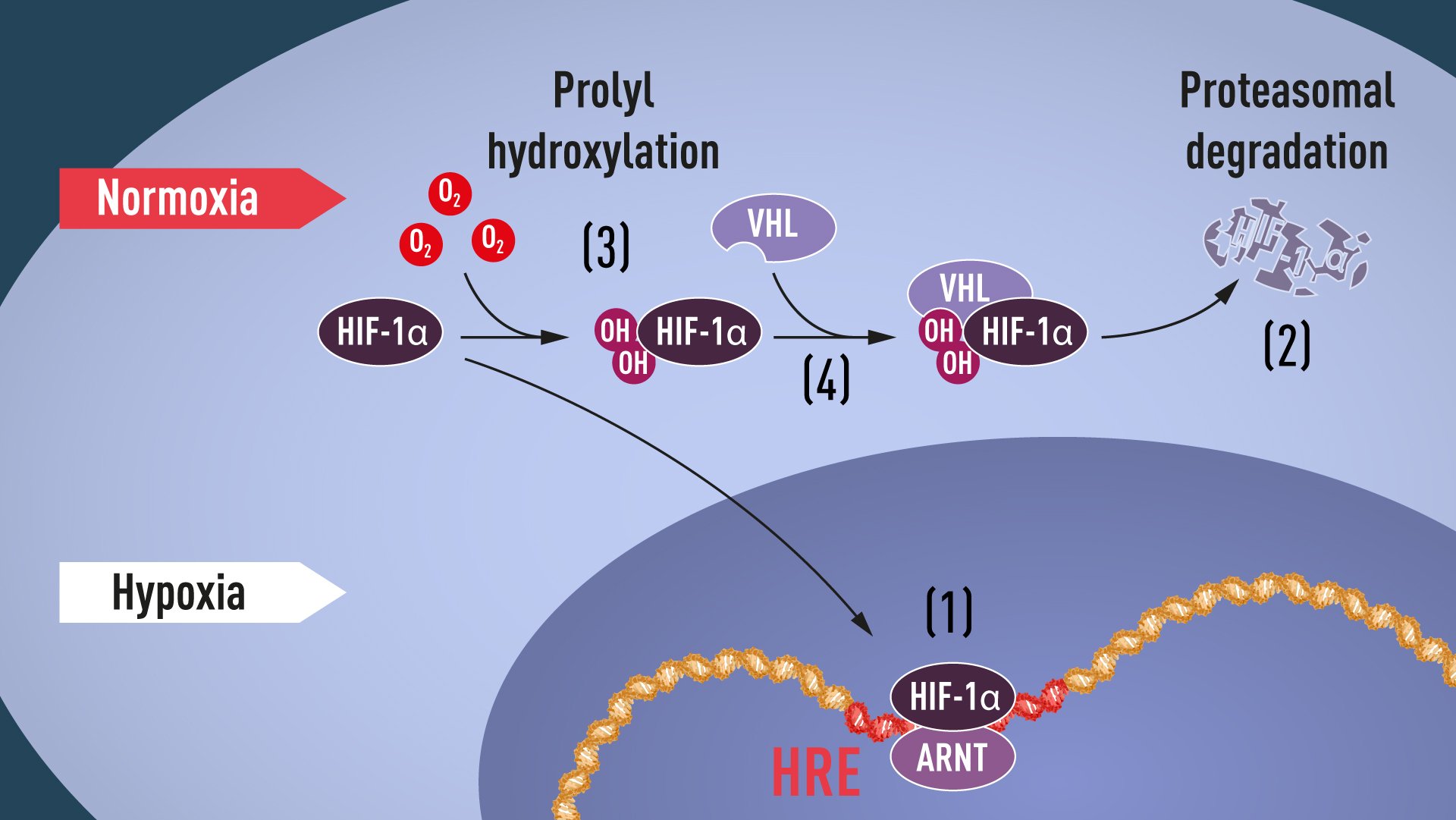
Figure 1. When oxygen levels are low (hypoxia), HIF-1α is protected from degradation and accumulates in the nucleus, where it associates with ARNT and binds to specific DNA sequences (HRE) in hypoxia-regulated genes (1). At normal oxygen levels, HIF-1α is rapidly degraded by the proteasome (2). Oxygen regulates the degradation process by the addition of hydroxyl groups (OH) to HIF-1α (3). The VHL protein can then recognize and form a complex with HIF-1α leading to its degradation in an oxygen-dependent manner (4).
Ratcliffe’s group and McKnight’s group independently identified the prolyl hydroxylase (PHD) genes involved in hydroxylating HIF-1α and EPAS1; these papers, describing the genetic isolation of the PHDs, were published in 2001 (Bruick and McKnight, 2001; Epstein et al., 2001). The Kaelin group also isolated the PHD genes, in their case using biochemical methods, and published that work in 2002 (Ivan et al., 2002). The identification of these hydroxylases gave rise to the possibility of creating specific PHD inhibitors to increase HIF activity; e.g., to increase EPO levels in patients with anemia.
A second oxygen-dependent mechanism, this time not for HIF-1α degradation but for inhibition of its activity as a transcription factor was discovered in 2001. Semenza and his group were the first to identify the factor involved, termed FIH-1 (for “factor inhibiting HIF”) (Mahon et al., 2001). FIH is also an oxygen-dependent hydroxylase, in this case one that hydroxylates an asparagine residue in the N-terminal activation domain (NTAD) of HIF-1α and EPAS1; this hydroxylation was found by Murray Whitelaw and Richard Bruick to interfere with the recruitment of the p300 transcriptional co-activator (Lando et al., 2002a; Lando et al., 2002b). In this way, oxygen not only promotes HIF-1α degradation via prolyl-hydroxylation of its ODD domain, but also can inhibit the transcriptional function of any HIF-1α or EPAS1 that has escaped VHL-dependent degradation. Thus, HIF activity has not one, but two independent mechanisms for oxygen-dependent post-translational inhibition. This indicates that keeping HIF levels properly and exactly regulated by cellular oxygen levels is necessarily a very finely tuned process.
The wide significance of the HIF pathway of control
Work by many groups have since shown the robustness of the HIF pathway, and its central role in modulating oxygen-influenced gene expression. Semenza, Ratcliffe, and Kaelin have remained central figures in this work since their original pathfinding discoveries. They have been involved in the further elucidation of the molecular biology of the HIF pathway, and have as well increased our understanding of the physiological roles played by hypoxic response in health and disease.
The discovery of the proline hydroxylases that regulate HIF-1α stability enabled a search for hydroxylase inhibitors to increase HIF levels; and this has now opened up new pathways for pharmacologic discovery (Giaccia et al., 2003). In fact, a number of potential drugs that increase HIF function by inhibiting the PHD enzymes are already far along in clinical trials, with a recent series of publications demonstrating their clinical efficacy in treatment of anemia (Chen et al., 2019a; Chen et al., 2019b).
Future applications to inhibit the HIF pathway are also on the horizon; these are envisioned as a means to slow the progression of some cancers that are induced by VHL mutations. One of these is a specific blocker of EPAS1 function that was recently described by Kaelin and colleagues as capable of slowing tumor growth of VHL mutant cells in animal models (Cho et al., 2016).
Pharmacologically increased HIF function may aid in the treatment of a wide range of diseases, as HIF has been shown to be essential for phenomena as diverse as immune function, cartilage formation, and wound healing. Conversely, inhibition of HIF function could also have many applications: increased levels of HIF are seen in many cancers as well as in some cardiovascular diseases, including stroke, heart attack, and pulmonary hypertension. It is thus likely that we are only at the beginning of applications of these Nobel Prize-winning discoveries, since it is clear that the response to oxygen in cells, tissues and organisms is one of the most central and important physiological adaptations that animals have.
References
Beck, I., Ramirez, S., Weinmann, R., and Caro, J. (1991). Enhancer element at the 3’-flanking region controls transcriptional response to hypoxia in the human erythropoietin gene. J Biol Chem 266, 15563-15566.
Bert, P. (1878). La Pression Barométrique: Recherches de Physiologie Expérimentale. (Paris: G. Masson).
Bert, P. (1882). Sur la richesse en hemoglobine du sang des animaux vivant sur les haut lieux. Comptes rendus de l’Académie des Sciences 94, 805-807.
Bondurant, M.C., and Koury, M.J. (1986). Anemia induces accumulation of erythropoietin mRNA in the kidney and liver. Mol Cell Biol 6, 2731-2733.
Bruick, R.K., and McKnight, S.L. (2001). A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337-1340.
Chen, N., Hao, C., Liu, B.C., Lin, H., Wang, C., Xing, C., Liang, X., Jiang, G., Liu, Z., Li, X., et al. (2019a). Roxadustat Treatment for Anemia in Patients Undergoing Long-Term Dialysis. N Engl J Med 381, 1011-1022.
Chen, N., Hao, C., Peng, X., Lin, H., Yin, A., Hao, L., Tao, Y., Liang, X., Liu, Z., Xing, C., et al. (2019b). Roxadustat for Anemia in Patients with Kidney Disease Not Receiving Dialysis. N Engl J Med 381, 1001-1010.
Cho, H., Du, X., Rizzi, J.P., Liberzon, E., Chakraborty, A.A., Gao, W., Carvo, I., Signoretti, S., Bruick, R.K., Josey, J.A., et al. (2016). On-target efficacy of a HIF-2alpha antagonist in preclinical kidney cancer models. Nature 539, 107-111.
Cockman, M.E., Masson, N., Mole, D.R., Jaakkola, P., Chang, G.W., Clifford, S.C., Maher, E.R., Pugh, C.W., Ratcliffe, P.J., and Maxwell, P.H. (2000). Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem 275, 25733-25741.
Compernolle, V., Brusselmans, K., Acker, T., Hoet, P., Tjwa, M., Beck, H., Plaisance, S., Dor, Y., Keshet, E., Lupu, F., et al. (2002). Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med 8, 702-710.
Duan, D.R., Pause, A., Burgess, W.H., Aso, T., Chen, D.Y., Garrett, K.P., Conaway, R.C., Conaway, J.W., Linehan, W.M., and Klausner, R.D. (1995). Inhibition of transcription elongation by the VHL tumor suppressor protein. Science 269, 1402-1406.
Ema, M., Taya, S., Yokotani, N., Sogawa, K., Matsuda, Y., and Fujii-Kuriyama, Y. (1997). A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A 94, 4273-4278.
Epstein, A.C., Gleadle, J.M., McNeill, L.A., Hewitson, K.S., O’Rourke, J., Mole, D.R., Mukherji, M., Metzen, E., Wilson, M.I., Dhanda, A., et al. (2001). C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43-54.
Fandrey, J. (2004). Oxygen-dependent and tissue-specific regulation of erythropoietin gene expression. Am J Physiol Regul Integr Comp Physiol 286, R977-988.
Flamme, I., Frohlich, T., von Reutern, M., Kappel, A., Damert, A., and Risau, W. (1997). HRF, a putative basic helix-loop-helix-PAS-domain transcription factor is closely related to hypoxia-inducible factor-1 alpha and developmentally expressed in blood vessels. Mech Dev 63, 51-60.
Giaccia, A., Siim, B.G., and Johnson, R.S. (2003). HIF-1 as a target for drug development. Nat Rev Drug Discov 2, 803-811.
Hogenesch, J.B., Chan, W.K., Jackiw, V.H., Brown, R.C., Gu, Y.Z., Pray-Grant, M., Perdew, G.H., and Bradfield, C.A. (1997). Characterization of a subset of the basic-helix-loop-helix-PAS superfamily that interacts with components of the dioxin signaling pathway. J Biol Chem 272, 8581-8593.
Huang, L.E., Gu, J., Schau, M., and Bunn, H.F. (1998a). Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A 95, 7987-7992.
Huang, L.E., Gu, J., Schau, M., and Bunn, H.F. (1998b). Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proceedings of the National Academy of Sciences of the United States of America 95, 7987-7992.
Iliopoulos, O., Kibel, A., Gray, S., and Kaelin, W.G., Jr. (1995). Tumour suppression by the human von Hippel-Lindau gene product. Nat Med 1, 822-826.
Iliopoulos, O., Levy, A.P., Jiang, C., Kaelin, W.G., Jr., and Goldberg, M.A. (1996). Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A 93, 10595-10599.
Ivan, M., Haberberger, T., Gervasi, D.C., Michelson, K.S., Gunzler, V., Kondo, K., Yang, H., Sorokina, I., Conaway, R.C., Conaway, J.W., et al. (2002). Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci U S A 99, 13459-13464.
Ivan, M., Kondo, K., Yang, H., Kim, W., Valiando, J., Ohh, M., Salic, A., Asara, J.M., Lane, W.S., and Kaelin, W.G., Jr. (2001). HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464-468.
Iyer, N.V., Kotch, L.E., Agani, F., Leung, S.W., Laughner, E., Wenger, R.H., Gassmann, M., Gearhart, J.D., Lawler, A.M., Yu, A.Y., et al. (1998). Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes & Development 12, 149-162.
Jaakkola, P., Mole, D.R., Tian, Y.M., Wilson, M.I., Gielbert, J., Gaskell, S.J., Kriegsheim, A., Hebestreit, H.F., Mukherji, M., Schofield, C.J., et al. (2001). Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468-472.
Jelkmann, W., and Hellwig-Burgel, T. (2001). Biology of erythropoietin. Adv Exp Med Biol 502, 169-187.
Kallio, P.J., Wilson, W.J., O’Brien, S., Makino, Y., and Poellinger, L. (1999). Regulation of the hypoxia-inducible transcription factor 1alpha by the ubiquitin-proteasome pathway. Journal of Biological Chemistry 274, 6519-6525.
Kamura, T., Sato, S., Iwai, K., Czyzyk-Krzeska, M., Conaway, R.C., and Conaway, J.W. (2000). Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci U S A 97, 10430-10435.
Kibel, A., Iliopoulos, O., DeCaprio, J.A., and Kaelin, W.G., Jr. (1995). Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science 269, 1444-1446.
Krieg, M., Haas, R., Brauch, H., Acker, T., Flamme, I., and Plate, K.H. (2000). Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 19, 5435-5443.
Lando, D., Peet, D.J., Gorman, J.J., Whelan, D.A., Whitelaw, M.L., and Bruick, R.K. (2002a). FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 16, 1466-1471.
Lando, D., Peet, D.J., Whelan, D.A., Gorman, J.J., and Whitelaw, M.L. (2002b). Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 295, 858-861.
Lavoisier, A. (1777). Mémoire sur la combustion en général. Mémoires de l’Académie des sciences, 592.
Lonergan, K.M., Iliopoulos, O., Ohh, M., Kamura, T., Conaway, R.C., Conaway, J.W., and Kaelin, W.G., Jr. (1998). Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol 18, 732-741.
Mahon, P.C., Hirota, K., and Semenza, G.L. (2001). FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev 15, 2675-2686.
Maxwell, P.H., Pugh, C.W., and Ratcliffe, P.J. (1993). Inducible operation of the erythropoietin 3’ enhancer in multiple cell lines: evidence for a widespread oxygen-sensing mechanism. Proc Natl Acad Sci U S A 90, 2423-2427.
Maxwell, P.H., Wiesener, M.S., Chang, G.W., Clifford, S.C., Vaux, E.C., Cockman, M.E., Wykoff, C.C., Pugh, C.W., Maher, E.R., and Ratcliffe, P.J. (1999). The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271-275.
Ohh, M., Park, C.W., Ivan, M., Hoffman, M.A., Kim, T.Y., Huang, L.E., Pavletich, N., Chau, V., and Kaelin, W.G. (2000). Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol 2, 423-427.
Pasteur, L. (1858). Nouveaux faits concernant l’histoire de la fermentation alcoolique. Comptes Rendus Chimie 47, 1011-1013.
Pause, A., Lee, S., Worrell, R.A., Chen, D.Y., Burgess, W.H., Linehan, W.M., and Klausner, R.D. (1997). The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci U S A 94, 2156-2161.
Peng, J., Zhang, L., Drysdale, L., and Fong, G.H. (2000). The transcription factor EPAS-1/hypoxia-inducible factor 2alpha plays an important role in vascular remodeling. Proc Natl Acad Sci U S A 97, 8386-8391.
Priestley, J. (1775). Experiments and observations on different kinds of air (London: Printed for J. Johnson, No. 72, in St. Paul’s Churchyard).
Pugh, C.W., O’Rourke, J.F., Nagao, M., Gleadle, J.M., and Ratcliffe, P.J. (1997). Activation of hypoxia-inducible factor-1; definition of regulatory domains within the alpha subunit. J Biol Chem 272, 11205-11214.
Pugh, C.W., Tan, C.C., Jones, R.W., and Ratcliffe, P.J. (1991). Functional analysis of an oxygen-regulated transcriptional enhancer lying 3’ to the mouse erythropoietin gene. Proc Natl Acad Sci U S A 88, 10553-10557.
Ryan, H.E., Lo, J., and Johnson, R.S. (1998). HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J 17, 3005-3015.
Salceda, S., and Caro, J. (1997). Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 272, 22642-22647.
Scheele, C. (1777). d. Königl. Schwed. Acad. d. Wissenschaft. Mitgliedes, Chemische Abhandlung von der Luft und dem Feuer. (Uppsala and Liepzig: Magnus Swederus, Buchhändler).
Schuster, S.J., Wilson, J.H., Erslev, A.J., and Caro, J. (1987). Physiologic regulation and tissue localization of renal erythropoietin messenger RNA. Blood 70, 316-318.
Semenza, G.L., Dureza, R.C., Traystman, M.D., Gearhart, J.D., and Antonarakis, S.E. (1990). Human erythropoietin gene expression in transgenic mice: multiple transcription initiation sites and cis-acting regulatory elements. Mol Cell Biol 10, 930-938.
Semenza, G.L., Koury, S.T., Nejfelt, M.K., Gearhart, J.D., and Antonarakis, S.E. (1991a). Cell-type-specific and hypoxia-inducible expression of the human erythropoietin gene in transgenic mice. Proc Natl Acad Sci U S A 88, 8725-8729.
Semenza, G.L., Nejfelt, M.K., Chi, S.M., and Antonarakis, S.E. (1991b). Hypoxia-inducible nuclear factors bind to an enhancer element located 3’ to the human erythropoietin gene. Proc Natl Acad Sci U S A 88, 5680-5684.
Semenza, G.L., Traystman, M.D., Gearhart, J.D., and Antonarakis, S.E. (1989). Polycythemia in transgenic mice expressing the human erythropoietin gene. Proc Natl Acad Sci U S A 86, 2301-2305.
Semenza, G.L., and Wang, G.L. (1992). A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 12, 5447-5454.
Srinivas, V., Zhang, L.P., Zhu, X.H., and Caro, J. (1999). Characterization of an oxygen/redox-dependent degradation domain of hypoxia-inducible factor alpha (HIF-alpha) proteins. Biochem Biophys Res Commun 260, 557-561.
Tanimoto, K., Makino, Y., Pereira, T., and Poellinger, L. (2000). Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO Journal 19, 4298-4309.
Tian, H., Hammer, R.E., Matsumoto, A.M., Russell, D.W., and McKnight, S.L. (1998). The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev 12, 3320-3324.
Tian, H., McKnight, S.L., and Russell, D.W. (1997). Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev 11, 72-82.
Wang, G.L., Jiang, B.H., Rue, E.A., and Semenza, G.L. (1995). Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proceedings of the National Academy of Sciences of the United States of America 92, 5510-5514.
Wang, G.L., and Semenza, G.L. (1993). General involvement of hypoxia-inducible factor 1 in transcriptional response to hypoxia. Proc Natl Acad Sci U S A 90, 4304-4308.
Wang, G.L., and Semenza, G.L. (1995). Purification and characterization of hypoxia-inducible factor 1. Journal of Biological Chemistry 270, 1230-1237.
Randall S. Johnson
Professor of Hypoxia Biology, Karolinska Institutet
Professor of Molecular Physiology and Pathology, University of Cambridge
Member of the Nobel Assembly
Karolinska Institutet, Stockholm, October 7, 2019
Correspondence: [email protected]
Illustration: Mattias Karlén
The Nobel Assembly, consisting of 50 professors at Karolinska Institutet, awards the Nobel Prize in Physiology or Medicine. Its Nobel Committee evaluates the nominations. Since 1901 the Nobel Prize has been awarded to scientists who have made the most important discoveries for the benefit of humankind.
Nobel Prize® is the registered trademark of the Nobel Foundation
Nobel Prizes and laureates
See them all presented here.

