Advanced information
Scientific background
Discoveries concerning nucleoside base modifications that enabled the development of effective mRNA vaccines against COVID-19
When SARS-CoV-2 emerged in late 2019 and rapidly spread to all parts of the world, few thought that vaccines could be developed in time to help curb the increasing global disease burden. Yet, several vaccines were approved in record time, with two of the fastest approved and most effective vaccines produced with the new mRNA technology. The concept of using mRNA for vaccination and in vivo delivery of therapeutic proteins was first proposed over 30 years ago, but several hurdles had to be overcome to make this a clinical reality. Early experiments demonstrated that in vitro transcribed mRNA stimulates undesired inflammatory responses and inefficient protein production in cells and tissues. A turning point was the discovery by Karikó and Weissman demonstrating that mRNA produced with modified nucleoside bases evades innate immune recognition and improves protein expression. These findings, combined with the development of efficient systems for in vivo mRNA delivery, stabilization of the SARS-CoV-2 spike antigen, and unparalleled investments by industry and governments, led to the approval of two highly successful mRNA-based COVID-19 vaccines in late 2020. The discovery by Karikó and Weissman was critical for making the mRNA vaccine platform suitable for clinical use at a time when it was most needed, making this an extraordinary contribution to medicine and paving the way for future mRNA applications.form suitable for clinical use at a time when it was most needed, making this an extraordinary contribution to medicine and paving the way for future mRNA applications.
In today’s globally interconnected society the risk of new pandemics is greater than ever before. Pandemics are usually caused by zoonotic viruses that cross the species barrier into humans and spread through droplet- or aerosol-mediated transmission, causing airway infections. Developing and deploying vaccines rapidly enough to mitigate an ongoing pandemic is an enormous challenge that had never been met before the COVID-19 pandemic. The rapid sharing of the SARS-CoV-2 genome sequence, along with extensive prior developments in molecular bio-logy, vaccine research, and drug delivery over the past several decades spurred unprecedented activity among vaccine researchers during 2020. Scientists in academia and industry launched projects in record time, with financial and logistical backing from governments, industry, and non-profit organizations. The new mRNA vaccine platform represented one of the most interesting options, but how well it would work against this new virus was unknown. No mRNA-based vaccine had been approved for human use before.
Virus vaccine platforms prior to COVID-19
Most licensed anti-viral vaccines available today are produced with traditional techniques based on weakened or inactivated whole viruses (Figure 1). Live attenuated virus vaccines, such as the combined rubella-mumps-measles vaccine and the yellow fever virus vaccine, induce robust and long-lived antibody and T cell-mediated immunity. For the development of the yellow fever virus vaccine, Max Theiler was awarded the Nobel Prize in Physiology or Medicine in 1951. Vaccines based on inactivated viruses, such as the tick-borne encephalitis vaccine and the hepatitis A vaccine, induce effective but more transient immune responses, requiring repeated boosting. With the revolution of molecular biology and the development of technologies for recombinant protein production, opportunities for more targeted vaccine approaches arose. The first vaccine produced using this approach was the hepatitis B vaccine (HBV), approved in 1986, which was followed by the approval of the first human papillomavirus (HPV) vaccine in 2006. The HBV and HPV vaccines contain single protein components of the respective virus and are referred to as subunit vaccines. These vaccines protect against virus-induced cancers and are life-saving success stories [1]. Developments in molecular biology also allowed the engineering of carrier viruses encoding heterologous antigens of interest. Such viral vectors efficiently enter cells where the encoded antigens are produced by the endogenous protein synthesis machinery. The first example of a licensed viral vector vaccine was the Vesicular stomatitis virus-based vaccine against Ebola, approved in 2019, which was soon followed by an adenovirus-based Ebola vaccine [2].
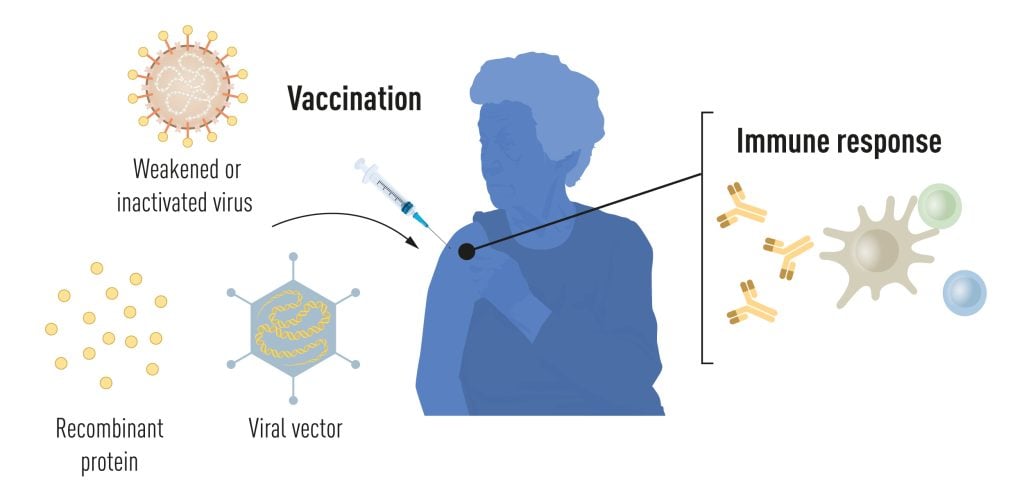
Currently used vaccines are made from weakened or inactivated whole viruses, recombinant viral protein components (subunit vaccines), or viral vectors delivering antigens of interest (vector vaccines). The vaccination event stimulates antigen-specific immune responses, which provide protection if the vaccinated person is later exposed to the live pathogen. © The Nobel Committee for Physiology or Medicine. Ill. Mattias Karlén
Both traditional whole virus-based vaccines and viral vector-based vaccines require cell culture-based manufacturing facilities. Vaccine researchers have therefore long been interested in the development of subunit vaccines that circumvent the need for large scale cell cultures by delivering nucleic acid (DNA or mRNA) directly to vaccine recipients, exploiting the body’s own capacity to produce proteins. There was a strong sentiment that the availability of such platforms would not only increase the world’s capacity to make vaccines, but also facilitate more rapid and less costly vaccine production in response to pandemics.
Early work on nucleic acid- and viral vector-based vaccines
The first demonstrations that nucleic acid-based immunizations could work date back to the early 1990´s when DNA vaccines [3] and mRNA vaccines [4] were first tested in mice. There were several potential advantages with these approaches. Not only are nucleic acid-based vaccines easy to manufacture; they are also flexible since the sequence can be easily changed to encode different antigens. Together with the ease of production, this makes iterative testing of new candidate vaccines and the generation of updated vaccines rapid and efficient. A biological advantage is that in addition to antibody and major histocompatibility complex (MHC) class II-restricted CD4+ T cell responses, which are also induced by other vaccine types, viral vector- and nucleic acid-based vaccines have the potential to stimulate cytotoxic CD8+ T cell responses since they allow presentation of endogenously produced antigenic peptides on MHC class I molecules. Induction of CD8+ T cells is particularly interesting in the context of cancer vaccines where the aim is to kill targeted tumor cells, and also for anti-viral vaccines aimed to eliminate infected cells. However, despite the potential advantages of nucleic acid-based vaccines, whether they would be well-tolerated and stimulate sufficiently robust immune response in humans to represent a viable path forward for clinical vaccine development was unclear.
Initially, DNA vaccines were considered more promising than mRNA vaccines since DNA is more stable. However, progress was slow and early encouraging results with DNA vaccines in small animals did not translate to humans [5]. A likely reason is that injected DNA must cross two barriers, the plasma membrane and the nuclear membrane, to reach the cellular compartment where transcription takes place (DNA conversion to mRNA). In contrast, mRNA-based vaccines only need to gain access the cell cytoplasm where translation takes place (mRNA conversion to protein), making delivery easier. An additional advantage with mRNA vaccines is that the delivered nucleic acid cannot integrate into the host genome, adding an important safety aspect to this platform. Despite these advantages, skepticism about the usefulness of the approach remained high since mRNA was considered too unstable for medical applications.
Against this background, the vaccine field turned to the use of engineered viral vectors as these have their own intrinsic mechanisms to enter cells and deliver genetic cargo. Since the 1990s, many different types of viral vector-based vaccines against a variety of pathogens have been tested preclinically, demonstrating both promising results and setbacks [6]. A drawback of viral vector-based vaccines is that in addition to the desired responses elicited against the antigen of interest, antibodies against the structural proteins used to package the vector may be induced, compromising booster responses if the same vector is used again. Nevertheless, effective viral vector-based vaccines using different types of engineered adenoviruses were developed during the COVID-19 pandemic and administered at scale, demonstrating their usefulness, especially in the early phase of a pandemic [7, 8].
During the 1990s, a small community of investigators continued to explore the use of mRNA as a potential vaccine platform. Early studies had demonstrated that mRNA purified from cells was translated into protein when reintroduced into oocytes [9]. Delivery into tissue of a living organism was the next challenge. The first study to demonstrate that injection of naked mRNA into skeletal muscle resulted in protein production in vivo was published by Philip Felgner and colleagues in 1990 [10]. Soon thereafter, Martinon et al. demonstrated the induction of antigen-specific cytotoxic T lymphocyte responses in mice injected with liposome-formulated mRNA encoding the influenza virus nucleoprotein [4].
In parallel, several investigators developed alphavirus replicon vaccines, which have the added advantage that a higher copy number of antigen-encoding transcripts are produced in each cell, resulting in the induction of robust antigen-specific immune responses following in vivo delivery of naked mRNA [11, 12]. These early studies stimulated the field and led to the demonstration of promising results in animal models, but it would take more than two decades until the first mRNA-based vaccine against an infection was tested in human clinical trials.
The discovery of mRNA and systems for in vitro transcription
To explore the potential of mRNA-based applications, an efficient system for mRNA production and manipulation was needed. For this, the field relied on a series of fundamental research discoveries starting in the 1950s. After the landmark discoveries of DNA as the inherited genetic material, the search started for the intermediate molecule that was transcribed from nuclear DNA and transported to the ribosomes in the cytoplasm to specify protein synthesis. Experiments on cells infected with the T2 bacteriophage identified a metabolically active RNA fraction constituting approximately 1% of the total cellular RNA [13] that had proper base ratios [14]. This unstable form of RNA, or messenger RNA (mRNA), was proposed to be the missing intermediate carrier of information [15], and the hypothesis soon gained experimental support through pulse-labeling experiments in bacteria [16, 17]. Around the same time, insight into how cells produce RNA from DNA was gained through the discovery of RNA polymerase [18-20]. In the following decades, several RNA polymerases were identified in bacteria and eukaryotic cells, including single-subunit RNA polymerases from the T7 [21] and SP6 [22] bacteriophages.
Building on the discovery of the more versatile bacteriophage RNA polymerases , Paul Krieg and Douglas Melton demonstrated that synthetic mRNA could be produced in large quantities in vitro by using the SP6 RNA polymerase and cDNA clones containing the SP6 promoter [23, 24]. Furthermore, the in vitro produced SP6 mRNA was efficiently translated into protein when injected into frog oocytes [23]. Around this time, the T7 RNA polymerase was cloned by William Studier’s lab [25] and developed into an efficient and inducible in vitro transcription system with a patent filed in 1984 [26]. The T7 RNA polymerase had several advantageous features, including highly specific binding to the T7 promoter (a conserved stretch of nucleotides -17 to +6 relative to the transcriptional start site) and an ability to transcribe RNA at a high speed. Similar efforts to harness the in vitro transcription capacity of T7 RNA polymerase were pursued [27]. The T7 in vitro transcription system became further optimized into a highly efficient cell-free system for large-scale production of any mRNA of interest, with major impact on science and biotechnology.
Delivering in vitro transcribed mRNA to cells and tissues
Another important research area focused on how to deliver nucleic acids into cells. An early strategy was to use liposomes, small cell-membrane-like vesicles composed of phospholipids and cholesterol. Already in 1978, researchers had described successful attempts at delivering purified globin mRNA into mouse lymphocytes and human epithelial cells using liposomes [28, 29] simply by trapping the mRNA inside the liposome vesicles. The field of nucleic acid de-livery improved thanks to the pioneering work by Philip Felgner while at Syntex Research. Felgner synthesized the first cationic lipid (DOTMA) and showed that it could form stable liposomes with nucleic acids [30]. The positively charged lipids improved both the entrapment of negatively charged nucleic acids (through electrostatic inter-actions) and fusion to the negatively charged cell membranes, resulting in improved delivery into cells. Cationic lipid-based liposomes (lipofectin) opened the door to the field of engineered DNA and RNA delivery into cells. Lipofectin was soon used to deliver in vitro transcribed mRNA into cultured cells to demonstrate protein production [31], encouraging future therapeutic applications. However, in vivo applications of lipofectin showed unwanted side effects and researchers continued the search for improved delivery systems.
A second major improvement was made in the lab of Pieter Cullis at the University of British Columbia with the development of ionizable cationic lipids. These lipids could be maintained in a positively charged or neutral form depending on the pH of the environment. Forming these lipid nano-particles (LNPs) at low pH had the benefits of cationic lipids in efficiently entrapping negatively charged mRNA within the vesicles. However, when delivered in vivo and exposed to physiological pH, the lipids lost their charge, which had several benefits including lower in vivo toxicity. The important discoveries by Cullis team spurred large industrial interest in the development of ionizable lipids. Notable, the delivery of nucleic acids was further optimized through the T-connector that could generate dense lipid nano-particles made of four components: an i) ionizable cationic lipid, ii) a helper lipid, iii) cholesterol and iv) polyethenylene glycol (PEG) [32]. More efficient ionizable cationic lipids were identified in large-scale screening programs in several biotech companies. Consequently, lipid nanoparticles now enable safe and efficient in vivo delivery of nucleic acids, including mRNA, into human cells. This advance is of great importance for clinical applications of nucleic acid-based technologies.
A vision to use mRNA for the delivery of therapeutic proteins
The potential of using the new molecular biology techniques to create mRNA-based vaccines or to treat human diseases by delivering mRNA to replace defective genes with functional ones, or by overexpressing a therapeutic protein, stimulated an enormous interest. In 1992, Jirikowski et al. used mRNA injection for in vivo expression of vasopressin to treat diabetes insipidus in a rodent model [33]. Around this time, a Hungarian research scientist at the University of Pennsylvania, Katalin Karikó, experimented with different forms of RNA with the ambition to optimize expression of therapeutic proteins. Karikó completed her PhD at the Biological Research Center in Szeged in 1982. Following post-doctoral work at the Hungarian Academy of Sciences and subsequent research positions at Temple University in Philadelphia and at the Uniformed Services University of the Health Sciences in Bethesda, she set up her own group at the Department of Neurosurgery at the University of Pennsylvania in 1997. Karikó had a strong drive to advance the mRNA platform and she systematically investigated different components of in vitro transcribed mRNA to identify requirements for optimal protein expression in cells and tissues [34]. Among several findings, she demonstrated that lipofectin-complexed mRNA encoding luciferase, a reporter protein, could be delivered to the rat brain and she showed that expression was improved when a longer poly(A) tail was added to the mRNA 3′ end [35]. Encouraged by these results, Karikó continued her quest to make the mRNA platform suitable for clinical use.
mRNA delivery to dendritic cells and the role of innate sensing
In the late 1990s, Karikó teamed up with Drew Weissman, a physician scientist with an interest in basic immunology and vaccine development, who had joined the University of Pennsylvania in 1997. Weissman had received his MD and PhD degrees from Boston University in immunology and microbiology in 1987. After a residency period at Beth Israel Deaconess Medical Center at Harvard Medical School in Boston, he joined Anthony Fauci’s group at the National Institutes of Health (NIH) for a post-doctoral fellowship to investigate how the human immunodeficiency virus type 1 (HIV-1) interacts with target receptors on different types of immune cells. Having established his own group at the University of Pennsylvania, he focused increasingly on vaccine research and the use of dendritic cells to prime immune responses. Ralph Steinman was awarded a Nobel Prize in Physiology or Medicine for the discovery of dendritic cells in 2011. With Weissman’s back-ground in immunology and Karikó’s expertise in RNA biochemistry, the two scientists complemented each other well and shared a passion for exploiting the use of mRNA in medical applications.
Together, Karikó and Weissman tested whether in vitro transcribed mRNA could be delivered to dendritic cells to exploit their antigen-presentation potential. A major goal of Weissman was to develop a vaccine against HIV-1, a virus that causes chronic infections. This was an exceptional challenge given the extensive immune evasion properties of this virus, setting it apart from viruses that cause acute infections. Weissman was interested in using dendritic cells to prime antigen-specific T cells and had developed systems to culture dendritic cells and assess their activation and antigen presenting capacities. Dendritic cells have exquisite abilities to both sense pathogens and prime naïve T cells and thus they bridge the innate and adaptive immune systems [36]. Karikó and Weissman showed that dendritic cells pulsed with in vitro transcribed mRNA encoding the HIV-1 structural protein, Gag, stimulated primary CD4+ and CD8+ T cell responses in vitro [37]. The team also found that the process of mRNA loading resulted in DC activation and maturation [38], which initially was interpreted as a positive effect since activated dendritic cells are superior in T cell priming. The negative consequences of innate immune activation by in vitro transcribed mRNA were not fully appreciated at this point. Interestingly, and somewhat counterintuitively, this would turn out to be a critical factor for advancing mRNA-based vaccines.
The observation that dendritic cells were activated following uptake of in vitro transcribed mRNA led to critical questions about which signaling pathways were engaged? Dendritic cells express both surface and endosomal Toll-like receptors (TLRs), which recognize distinct molecular structures referred to as pathogen-associated molecular patterns (PAMPs) [39]. TLR binding to PAMPs results in intracellular signaling and pro-duction of anti-viral cytokines including type 1 interferons, an effective warning system to detect incoming pathogens. Studies of how TLRs distinguish different forms of nucleic acid had gained traction after Hemmi et al. showed that unmethylated CpG motifs, abundant in microbial but rare in mammalian DNA, activate TLR9 [40].
Within a few years, the ligands for most nucleic acid sensing TLRs had been identified, including TLR3 that senses double-stranded RNA (dsRNA), a viral replication intermediate, and TLR7 and TLR8 that sense single-stranded viral RNA and some forms of synthetic RNA [41, 42]. In 2004, Karikó and Weissman reported that in vitro transcribed mRNA contains dsRNA contaminants that can activate TLR3, leading to a cytokine response [43]. Another important clue was obtained when Koski, Karikó and Weissman together with Brian Czerniecki and colleagues demonstrated that transfection of dendritic cells with in vitro transcribed mRNA stimulated a cytokine response similar to that observed with prokaryotic RNA. Experimental manipulations to increase the poly(A) length of in vitro transcribed mRNA led to significantly reduced IL-12 production. However, this was not the full explanation for the observed effects. When four homopolynucleotides, polyuridylic acid (pU), polyguanylic acid (pG), polycytidylic acid (pC), polyadenylic acid (pA), were tested using IL-12 as a read-out for DC activation, only pU induced a response, suggesting that the nucleotide content also played a role [44]. A similar finding, using interferon alpha as a readout, was reported the same year from Reis e Sousa’s group in their studies of RNA recognition by TLR7 [41].
The Kariko, Weissman breakthrough
Karikó and Weissman continued their careful studies of different types of RNA and the work resulted in a breakthrough publication in 2005. The study described the influence of mRNA base modifications on the cytokine response by dendritic cells [45]. They showed that eukaryotic mRNA and tRNA, in which base modifications are abundant, did not stimulate a cytokine response while prokaryotic and in vitro-transcribed mRNA did. They further showed that the incorporation of pseudouridine (ψ), 5-methylcytidine (m5C), N6-methyladenosine (m6A), 5-methyluridine (m5U) or 2-thiouridine (s2U) into in vitro transcribed mRNA abrogated activation of inflammatory responses when these mRNAs were added to dendritic cells [45]. The incorporation of m6A and s2U almost completely abrogated recognition by TLR3, while TLR7 and TLR8 activation could be evaded using m6A, s2U, m5C, m5U and ψ. Importantly, only modifications of uridines (m5U, s2U and ψ) abolished DC activation (Figure 2).
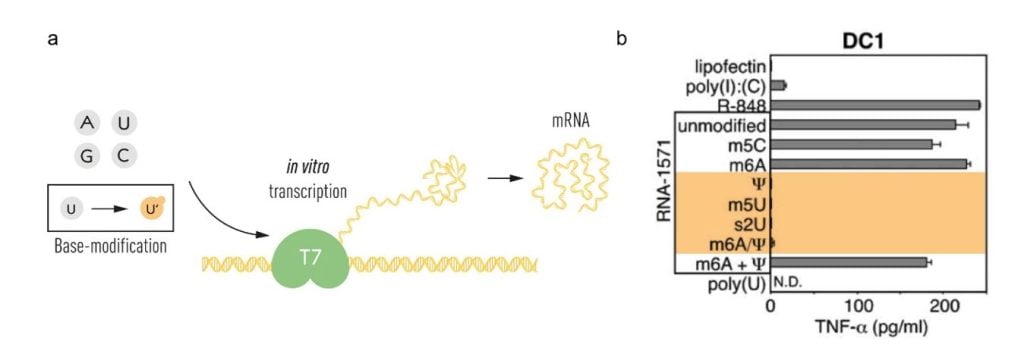
(a) The T7 in vitro transcription system was used to produce mRNA with canonical RNA bases (A, U, G and C) or modified bases. (b) The bases used for in vitro transcription of RNA-1571 are shown, with those that did not result in TNF-alpha secretion indicated in orange (modified from Karikó et al. Immunity 2005). © The Nobel Committee for Physiology or Medicine. Ill. Mattias Karlén
To date, researchers have uncovered more than one hundred different post-transcriptional modifications in RNA and shown that modifications are more extensive in RNA of eukaryotes than prokaryotes [46, 47]. Pseudouridine (Ψ) was discovered already in 1951 [48] and is one of the most abundant RNA modifications, initially found in tRNAs and small nuclear RNAs (snRNAs) and more recently in other types of RNA. Cells modify RNA through enzymatic reactions, for example pseudouridine is catalyzed by pseudouridine synthase enzymes, or using small ribonucleoprotein (snoRNPs) complexes. RNA modifications contribute to RNA stability, base-pairing specificity, folding and other functional properties. Of the over one hundred RNA modifications known [49], limited functional data exists on most modifications. Understanding the physiological implications of these modifications therefore remains an active research field.
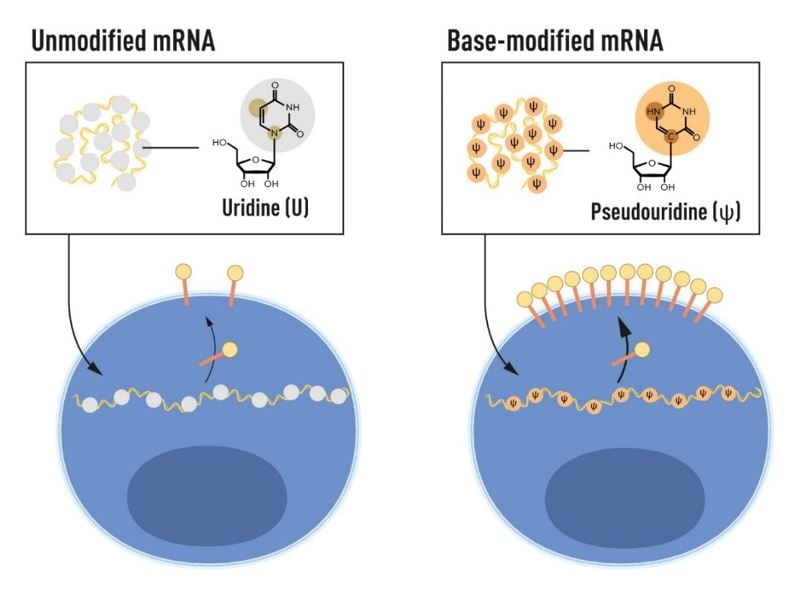
The Karikó and Weissman discovery explained an observation made over 40 years earlier by Isaacs and colleagues demonstrating that delivery of deaminated RNA into cells resulted in a stronger type 1 interferon response than control RNA [50]. Deamination increases the proportion of uridines in the RNA, which Kariko and Weissman had demonstrated was critical for DC activation. Later work showed that the use of N1-methylpseudo-uridine (m1ψ), alone or in combination with m5C, further improved the mRNA platform both in terms of reducing recognition of innate immune receptors and increasing protein expression [51], the latter was in part explained by an increased ribosome occupancy on m1ψ-containing mRNA [52]. Today, m1ψ is the most common modified base used in mRNA vaccine production, including in the two COVID-19 vaccines approved in late 2020, as discussed below.
Following their breakthrough discovery that incorporation of modified bases evades undesired immune activation by in vitro transcribed mRNA, Karikó and Weissman demonstrated that pseudouridine-containing mRNA was also more efficiently translated, resulting in higher protein production in cells that have taken up the mRNA [53] (Figure 3). In the same study, they showed that delivery of modified mRNA into the spleen of mice led to increased protein production and decreased immune activation, an important demonstration for future therapeutic applications. Karikó, Weissman and colleagues further demonstrated that in vitro transcribed mRNA activates protein kinase R (PKR), an anti-viral protein that protects cells from invading pathogens by recognizing dsRNA by phosphorylating the eukaryotic translation initiation factor 2 alpha (eIF2a), blocking protein translation. The team showed that the use of modified bases reduced activation of PKR and improved protein production [54]. Recognition of in vitro transcribed mRNA by the 2’5’ oligoadenylate synthetase (OAS) and degradation by the OAS-induced Rnase L enzyme were also decreased with RNA containing modified bases [55].
Furthermore, Karikó and colleagues showed that dsRNA contaminants produced during in vitro transcription could be removed through an HPLC purification step [56], or as later reported together with Uğur Şahin and colleagues at BioNTech, by using a cellulose-based purification step [57], further improving the expression of protein from in vitro transcribed mRNA.
Research leading up to the mRNA vaccines against COVID-19
By 2010, three main companies with programs focusing on the emerging mRNA technology had been established: CureVac, founded in 2000 aimed to develop vaccines against infections and cancer; BioNTech founded in 2008 had the objective to develop personalized cancer vaccines; and Moderna, founded in 2010 planned to use the mRNA platform to reprogram somatic cells to pluripotent cells and to deliver therapeutic proteins, for example to repair damaged tissue. All three companies collaborated closely with academic researchers to improve the technology and evaluate their respective platforms in disease areas of interest.
The team behind Curevac, including Ingmar Hoerr, Günter Jung, Steve Pascolo and Hans-Georg Rammensee, had realized the potential of the mRNA technology early on. They developed approaches to improve the efficiency of protein production through optimizations of the mRNA 5’ and 3′ untranslated regions and codon optimization, without using modified bases. In 2000, they reported that administration of RNA, either naked or liposome-complexed, induced antigen-specific adaptive immune responses in mice (antibody and CD8+ T cell responses) with the liposome-encapsulated RNA giving higher responses [58]. They evaluated their first mRNA vaccine in humans approximately eight years later when genetic material from tumors of melanoma patients was extracted and used to generate mRNA that was administered as an autologous vaccine with granulocyte-macrophage colony-stimulating factor (GM-CSF) as an adjuvant. The approach was shown to be safe and to increase anti-tumor immune responses in some patients [59]. In 2012, the Curevac team reported elicitation of protective immune responses against influenza virus infection in several animal models [60] and in 2017, the first mRNA-based vaccine against an infectious disease, rabies, was tested in clinical trials.
Activities in the mRNA vaccine field now expanded rapidly. In 2017, promising pre-clinical results of mRNA-based Zika virus vaccines that used modified bases were reported by Norbert Pardi and Weissman [61] and by Michael Diamond and colleagues at Washington University School of Medicine [62]. The latter study, which described vaccination of pregnant females, demonstrated protection against viral transmission to the fetus, a major concern with Zika virus infections. In 2017, Moderna announced the start of a clinical trial with an mRNA-based vaccine against Zika virus (ClinicalTrials.gov: NCT03014089). Moderna also initiated two phase I clinical trials to evaluate the safety and immunogenicity of their mRNA vaccine candidates against influenza virus H10N8 and H7N9, two avian influenza strains with pandemic potential [63, 64] (ClinicalTrials.gov NCT03076385 and NCT03345043).
Around the time of the Zika vaccine trial, Moderna also initiated collaborations with Barney Graham and his team at the Vaccine Research Center at the NIH to develop an mRNA-based vaccine against Middle East Respiratory Syndrome coronavirus (MERS-CoV). The vaccine encoded a prefusion-stabilized form of the MERS spike where, among other modifications, prolines were introduced in the S2 domain to prevent the metastable prefusion form transitioning into the post-fusion form [65]. Early work by Qiao et al. had showed that the introduction of prolines in the influenza virus hemagglutinin 2 domain (HA2), which undergoes a loop to helix transition at low pH, interferes with the ability of the influenza virus to fuse with host membranes [66]. Based on this finding, and the knowledge that viruses from different families have evolved similar solutions for fusing with target cells, appropriately positioned prolines have been substituted into the spike glycoproteins of several viruses to stabilize them in their respective prefusion forms, including but not limited to HIV-1 [67], Respiratory syncytial virus [68] and SARS-CoV-2 [69]. The high-resolution structure of the SARS-CoV-2 spike published in record time by Jason McLellan’s group in early 2020 proved invaluable for several of the successful COVID-19 vaccines, as well as for the definition of neutralizing antibody epitopes and antibody escape mutations in later emerging SARS-CoV-2 variants, information that is of great importance for our understanding of vaccine-induced immune protection. The prefusion-stabilized form of the SARS-CoV-2 spike was used in the mRNA vaccines developed by Pfizer/BioNTech and Moderna (Figure 4), as well as in the vector vaccine by Janssen and the protein-based vaccine developed by Novavax.
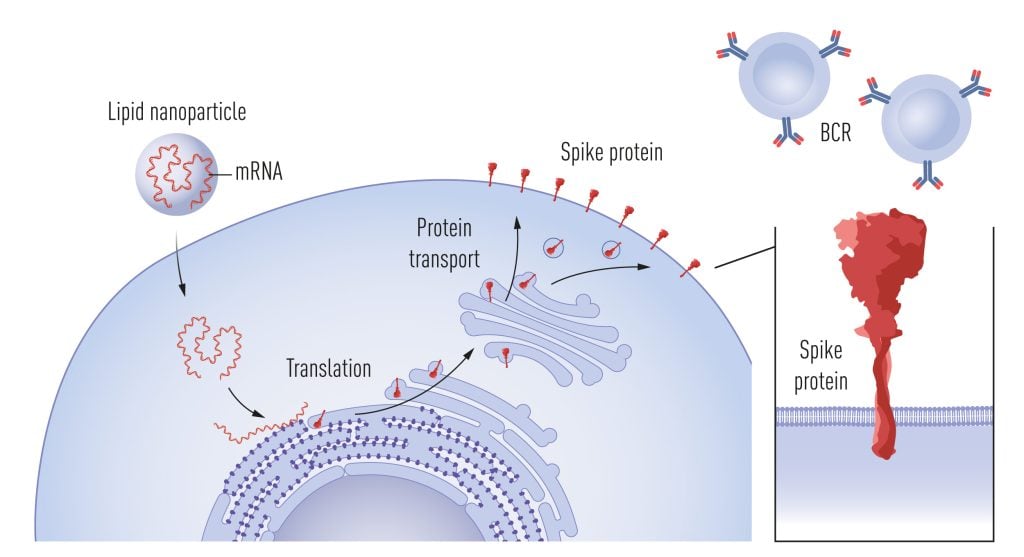
Following uptake of mRNA into cells, facilitated by lipid nanoparticles, the mRNA acts as a template for spike protein production. Spike is then transiently expressed on the cell surface, where it is recognized by B cells via their B cell receptors (BCRs), stimulating the secretion of spike-specific antibodies. © The Nobel Committee for Physiology or Medicine. Ill. Mattias Karlén
The moment was ripe for mRNA vaccines
When the pandemic broke in early 2020, mRNA companies acted quickly to develop COVID-19 vaccines. BioNTech and Moderna chose to use mRNA with modified bases building on the discoveries by Karikó and Weissmann. BioNTech, with Uğur Şahin and Özlem Türeci in the lead, worked in partnership with Pfizer [70, 71], while Moderna collaborated closely with the VRC/NIH where Barney Graham and an assembled team performed the vaccine evaluation [72-74]. Undoubtedly, the SARS-CoV-2 pandemic was a decisive event that led to large-scale investments in the mRNA vaccine technology, including the design of clinical trials that ran in parallel rather than sequentially, shortening the time required for clinical trials considerably while still completing all the necessary steps [75]. The collective funding and support from governments, international organizations, and industry resulted in the completion of vaccine safety and efficacy trials in record time with both the Pfizer/BioNTech’s and Moderna’s mRNA vaccinesgaining approval within a year of the SARS-CoV-2 outbreak. This development was made possible thanks to decades of basic research and optimization of the mRNA platforms, as reviewed in [76]. Both the Pfizer/BioNTech’s and Moderna’s mRNA vaccines had complete substitutions of uridine with N1-methylpseudouridine (m1ψ) to avoid unwanted inflammatory responses, to ramp up protein translation, and to enable higher mRNA amounts to be used in each vaccine dose.
The phase 3 trials, which were based on results obtained after two mRNA vaccinations, showed that the level of protection against symptomatic COVID-19 was very high, 95% efficacy for Pfizer/BioNTech’s vaccine (Polack 2020) and 94% for Moderna’s vaccine (Baden 2021). Both vaccines induced potent antibody responses, as well as memory B cell and T cell responses, providing protection against severe disease and death. Follow-up studies showed that the serological responses were relatively short-lived, and the research community soon showed that additional booster immunizations greatly improved protection, especially against the more infectious Omicron variant [77]. The spread of new SARS-CoV-2 variants is well documented and several Omicron subvariants are in circulation. The global scientific community continues to monitor the virus evolution to track the emergence of new variants and guide the design of updated vaccines. The past year has demonstrated that the mRNA platform is amenable to the production of updated vaccines at a speed that is currently not matched by other vaccine platforms.
Several COVID-19 vaccines have contributed to saving lives and reducing unsustainable pressures on health care systems since 2021. The mRNA technology represents a critical addition to the arsenal of platforms that can be used in vaccine production, not the least in response to pandemics when scalability and flexibility are essential. The wide-spread use of the two COVID-19 mRNA vaccines over the past years demonstrates the significant potential of the technology and shows that serious adverse effects to the two licensed mRNA vaccines were exceptionally rare [78], providing a strong foundation for future applications.
Are modified bases required for all clinical mRNA applications?
Several applications of the mRNA platform are now in development, including for vaccines against infections and cancer and for the delivery of therapeutic or immunomodulatory proteins. Different mRNA applications may have different requirements for modified bases. For prophylactic vaccines that are given to large numbers of healthy individuals, reactogenicity to the injection is an important consideration. A mild transient reaction may be acceptable if it is limited to the injection site, while systemic inflammatory symptoms such as fever, myalgia and headaches are undesired or, depending on the severity, unacceptable. The acceptable level of reactogenicity must be decided for each specific vaccine product, and this depends on the magnitude of the benefit of inducing a protective response. Thus, striking the right dose balance between reactogenicity and efficacy for a given vaccine can be challenging [79].
Khoury et al. reported that the protective effects of all COVID-19 vaccines for which results were available by mid-2021 correlated with the mean neutralizing antibody titers against the founder virus elicited in each of the trials [80]. This aligns with data from other licensed anti-viral vaccines for which protection against disease is known to correlate with neutralizing antibodies [81]. Once the results obtained in Curevac’s clinical trial were available [82] they were compared with the results from the other trials [83]. This analysis showed that the neutralizing antibody titers elicited by the Curevac vaccine were lower than those elicited by Pfizer/BioNTech’s and Moderna’s mRNA vaccines, suggesting that the lower mRNA dose used in the Curevac trial compromised the protective effect of this vaccine. A definitive comparison of the different mRNA vaccines is confounded by the fact that more neutralization-resistant variants were circulating by the time Curevac ran its phase 3 trial. However, the results support that the use of base-modifications in in vitro transcribed mRNA encoding the SARS-CoV-2 spike was critical for the development of mRNA-vaccines that could be given at sufficiently high doses to protect against COVID-19.
In the future, additional approaches to optimize mRNA for the development of clinically useful products, such as circular RNA [84], replicons [85] and other types of RNA that do not contain base-modifications, will likely be developed. Recent clinical studies with therapeutic mRNA vaccination combined with checkpoint inhibition using mutated or unmutated tumor antigens demonstrate successful induction of tumor-specific T cell responses in melanoma and pancreatic ductal adenocarcinoma patients [86-88]. The mRNA used in these trials used unmodified bases but included modifications to the poly(A) tail described to increase mRNA stability and translational efficiency [89]. Thus, alternative approaches to generate effective mRNA-based vaccines and therapeutics are in development. There are now numerous clinical trials using different forms of mRNA to induce prophylactic or therapeutic responses in the fields of infection [90] and cancer [91, 92], and this is predicted to increase over the coming years.
Summary
The approval of two effective and safe COVID-19 mRNA-vaccines in late 2020 propelled the mRNA vaccine field into a new era. The discovery that the use of modified bases in in vitro-transcribed mRNA circumvents undesired inflammatory responses and increases protein production following delivery to cells demonstrates the value of basic research. The results published by Karikó and Weissman in their seminal 2005 paper received little attention at the time but laid the foundation for critically important developments that have served humanity during the COVID-19 pandemic.
Gunilla Karlsson Hedestam, PhD, Professor at Karolinska Institutet ([email protected]), Member of the Nobel Committee
Rickard Sandberg, PhD, Professor at Karolinska Institutet ([email protected]), Member of the Nobel Committee
Illustrations: Mattias Karlén
References
1. Stanley, M., Tumour virus vaccines: hepatitis B virus and human papillomavirus. Philos Trans R Soc Lond B Biol Sci, 2017. 372(1732).
2. Woolsey, C. and T.W. Geisbert, Current state of Ebola virus vaccines: A snapshot. PLoS Pathog, 2021. 17(12): p. e1010078.
3. Tang, D.C., M. DeVit, and S.A. Johnston, Genetic immunization is a simple method for eliciting an immune response. Nature, 1992. 356(6365): p. 152-4.
4. Martinon, F., et al., Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur J Immunol, 1993. 23(7): p. 1719-22.
5. Liu, M.A. and J.B. Ulmer, Human clinical trials of plasmid DNA vaccines. Adv Genet, 2005. 55: p. 25-40.
6. Draper, S.J. and J.L. Heeney, Viruses as vaccine vectors for infectious diseases and cancer. Nat Rev Microbiol, 2010. 8(1): p. 62-73.
7. Falsey, A.R., et al., Phase 3 Safety and Efficacy of AZD1222 (ChAdOx1 nCoV-19) Covid-19 Vaccine. N Engl J Med, 2021. 385(25): p. 2348-2360.
8. Sadoff, J., et al., Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against Covid-19. N Engl J Med, 2021. 384(23): p. 2187-2201.
9. Gurdon, J.B., et al., Use of frog eggs and oocytes for the study of messenger RNA and its translation in living cells. Nature, 1971. 233(5316): p. 177-82.
10. Wolff, J.A., et al., Direct gene transfer into mouse muscle in vivo. Science, 1990. 247(4949 Pt 1): p. 1465-8.
11. Johanning, F.W., et al., A Sindbis virus mRNA polynucleotide vector achieves prolonged and high level heterologous gene expression in vivo. Nucleic Acids Res, 1995. 23(9): p. 1495-501.
12. Zhou, X., et al., Self-replicating Semliki Forest virus RNA as recombinant vaccine. Vaccine, 1994. 12(16): p. 1510-4.
13. Hershey, A.D., Nucleic acid economy in bacteria infected with bacteriophage T2. J Gen Physiol, 1953. 37(1): p. 1-23.
14. Volkin, E. and L. Astrachan, Phosphorus incorporation in Escherichia coli ribonucleic acid after infection with bacteriophage T2. Virology, 1956. 2(2): p. 149-61.
15. Jacob, F. and J. Monod, Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol, 1961. 3: p. 318-56.
16. Brenner, S., F. Jacob, and M. Meselson, An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature, 1961. 190: p. 576-581.
17. Gros, F., et al., Unstable ribonucleic acid revealed by pulse labelling of Escherichia coli. Nature, 1961. 190: p. 581-5.
18. Hurwitz, J., A. Bresler, and R. Diringer, The Enzymic Incorporation of Ribonucleotides into Polyribonucleotides and the Effect of DNA. Biochemical and Biophysical Research Communications, 1960. 3(1): p. 15-19.
19. Stevens, A., Incorporation of the Adenine Ribonucleotide into Rna by Cell Fractions from, E-Coli B. Biochemical and Biophysical Research Communications, 1960. 3(1): p. 92-96.
20. Weiss, S.B. and L. Gladstone, A Mammalian System for the Incorporation of Cytidine Triphosphate into Ribonucleic Acid. Journal of the American Chemical Society, 1959. 81(15): p. 4118-4119.
21. Chamberlin, M., J. McGrath, and L. Waskell, New RNA polymerase from Escherichia coli infected with bacteriophage T7. Nature, 1970. 228(5268): p. 227-31.
22. Butler, E.T. and M.J. Chamberlin, Bacteriophage SP6-specific RNA polymerase. I. Isolation and characterization of the enzyme. J Biol Chem, 1982. 257(10): p. 5772-8.
23. Krieg, P.A. and D.A. Melton, Functional messenger RNAs are produced by SP6 in vitro transcription of cloned cDNAs. Nucleic Acids Res, 1984. 12(18): p. 7057-70.
24. Melton, D.A., et al., Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Res, 1984. 12(18): p. 7035-56.
25. Dunn, J.J. and F.W. Studier, Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J Mol Biol, 1983. 166(4): p. 477-535.
26. Studier, F.W. and B.A. Moffatt, Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol, 1986. 189(1): p. 113-30.
27. Tabor, S. and C.C. Richardson, A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc Natl Acad Sci U S A, 1985. 82(4): p. 1074-8.
28. Dimitriadis, G.J., Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature, 1978. 274(5674): p. 923-4.
29. Ostro, M.J., et al., Evidence for translation of rabbit globin mRNA after liposome-mediated insertion into a human cell line. Nature, 1978. 274(5674): p. 921-3.
30. Felgner, P.L., et al., Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc Natl Acad Sci U S A, 1987. 84(21): p. 7413-7.
31. Malone, R.W., P.L. Felgner, and I.M. Verma, Cationic liposome-mediated RNA transfection. Proc Natl Acad Sci U S A, 1989. 86(16): p. 6077-81.
32. Jeffs, L.B., et al., A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA. Pharm Res, 2005. 22(3): p. 362-72.
33. Jirikowski, G.F., et al., Reversal of diabetes insipidus in Brattleboro rats: intrahypothalamic injection of vasopressin mRNA. Science, 1992. 255(5047): p. 996-8.
34. Kariko, K., A. Kuo, and E. Barnathan, Overexpression of urokinase receptor in mammalian cells following administration of the in vitro transcribed encoding mRNA. Gene Ther, 1999. 6(6): p. 1092-100.
35. Kariko, K., et al., In vivo protein expression from mRNA delivered into adult rat brain. J Neurosci Methods, 2001. 105(1): p. 77-86.
36. Steinman, R.M., The dendritic cell system and its role in immunogenicity. Annu Rev Immunol, 1991. 9: p. 271-96.
37. Weissman, D., et al., HIV gag mRNA transfection of dendritic cells (DC) delivers encoded antigen to MHC class I and II molecules, causes DC maturation, and induces a potent human in vitro primary immune response. J Immunol, 2000. 165(8): p. 4710-7.
38. Ni, H., et al., Extracellular mRNA induces dendritic cell activation by stimulating tumor necrosis factor-alpha secretion and signaling through a nucleotide receptor. J Biol Chem, 2002. 277(15): p. 12689-96.
39. Akira, S. and H. Hemmi, Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett, 2003. 85(2): p. 85-95.
40. Hemmi, H., et al., A Toll-like receptor recognizes bacterial DNA. Nature, 2000. 408(6813): p. 740-5.
41. Diebold, S.S., et al., Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science, 2004. 303(5663): p. 1529-31.
42. Heil, F., et al., Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science, 2004. 303(5663): p. 1526-9.
43. Kariko, K., et al., mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem, 2004. 279(13): p. 12542-50.
44. Koski, G.K., et al., Cutting edge: innate immune system discriminates between RNA containing bacterial versus eukaryotic structural features that prime for high-level IL-12 secretion by dendritic cells. J Immunol, 2004. 172(7): p. 3989-93.
45. Kariko, K., et al., Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity, 2005. 23(2): p. 165-75.
46. Limbach, P.A., P.F. Crain, and J.A. McCloskey, Summary: the modified nucleosides of RNA. Nucleic Acids Res, 1994. 22(12): p. 2183-96.
47. Machnicka, M.A., et al., MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res, 2013. 41(Database issue): p. D262-7.
48. Cohn, W.E. and E. Volkin, Nucleoside-5′-Phosphates from Ribonucleic Acid. Nature, 1951. 167(4247): p. 483-484.
49. Rozenski, J., P.F. Crain, and J.A. McCloskey, The RNA Modification Database: 1999 update. Nucleic Acids Res, 1999. 27(1): p. 196-7.
50. Isaacs, A., R.A. Cox, and Z. Rotem, Foreign nucleic acids as the stimulus to make interferon. Lancet, 1963. 2(7299): p. 113-6.
51. Andries, O., et al., N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J Control Release, 2015. 217: p. 337-44.
52. Svitkin, Y.V., et al., N1-methyl-pseudouridine in mRNA enhances translation through eIF2alpha-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res, 2017. 45(10): p. 6023-6036.
53. Kariko, K., et al., Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther, 2008. 16(11): p. 1833-40.
54. Anderson, B.R., et al., Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res, 2010. 38(17): p. 5884-92.
55. Anderson, B.R., et al., Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res, 2011. 39(21): p. 9329-38.
56. Kariko, K., et al., Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res, 2011. 39(21): p. e142.
57. Baiersdorfer, M., et al., A Facile Method for the Removal of dsRNA Contaminant from In Vitro-Transcribed mRNA. Mol Ther Nucleic Acids, 2019. 15: p. 26-35.
58. Hoerr, I., et al., In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol, 2000. 30(1): p. 1-7.
59. Weide, B., et al., Results of the first phase I/II clinical vaccination trial with direct injection of mRNA. J Immunother, 2008. 31(2): p. 180-8.
60. Petsch, B., et al., Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat Biotechnol, 2012. 30(12): p. 1210-6.
61. Pardi, N., et al., Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature, 2017. 543(7644): p. 248-251.
62. Richner, J.M., et al., Vaccine Mediated Protection Against Zika Virus-Induced Congenital Disease. Cell, 2017. 170(2): p. 273-283 e12.
63. Bahl, K., et al., Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol Ther, 2017. 25(6): p. 1316-1327.
64. Feldman, R.A., et al., mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine, 2019. 37(25): p. 3326-3334.
65. Pallesen, J., et al., Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc Natl Acad Sci U S A, 2017. 114(35): p. E7348-E7357.
66. Qiao, H., et al., Specific single or double proline substitutions in the “spring-loaded” coiled-coil region of the influenza hemagglutinin impair or abolish membrane fusion activity. J Cell Biol, 1998. 141(6): p. 1335-47.
67. Sanders, R.W., et al., Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J Virol, 2002. 76(17): p. 8875-89.
68. Krarup, A., et al., A highly stable prefusion RSV F vaccine derived from structural analysis of the fusion mechanism. Nat Commun, 2015. 6: p. 8143.
69. Wrapp, D., et al., Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science, 2020. 367(6483): p. 1260-1263.
70. Mulligan, M.J., et al., Phase I/II study of COVID-19 RNA vaccine BNT162b1 in adults. Nature, 2020. 586(7830): p. 589-593.
71. Sahin, U., et al., COVID-19 vaccine BNT162b1 elicits human antibody and TH1 T cell responses. Nature, 2020. 586(7830): p. 594-599.
72. Corbett, K.S., et al., SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature, 2020. 586(7830): p. 567-571.
73. Francica, J.R., et al., Protective antibodies elicited by SARS-CoV-2 spike protein vaccination are boosted in the lung after challenge in nonhuman primates. Sci Transl Med, 2021. 13(607).
74. Jackson, L.A., et al., An mRNA Vaccine against SARS-CoV-2 – Preliminary Report. N Engl J Med, 2020. 383(20): p. 1920-1931.
75. Accelerating vaccine trials. Bull World Health Organ, 2021. 99(7): p. 482-483.
76. Sahin, U., K. Kariko, and O. Tureci, mRNA-based therapeutics–developing a new class of drugs. Nat Rev Drug Discov, 2014. 13(10): p. 759-80.
77. Garcia-Beltran, W.F., et al., mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell, 2022. 185(3): p. 457-466 e4.
78. Husby, A. and L. Kober, COVID-19 mRNA vaccination and myocarditis or pericarditis. Lancet, 2022. 399(10342): p. 2168-2169.
79. Lee, J., et al., Knife’s edge: Balancing immunogenicity and reactogenicity in mRNA vaccines. Exp Mol Med, 2023. 55(7): p. 1305-1313.
80. Khoury, D.S., et al., Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat Med, 2021. 27(7): p. 1205-1211.
81. Plotkin, S.A., Correlates of protection induced by vaccination. Clin Vaccine Immunol, 2010. 17(7): p. 1055-65.
82. Kremsner, P.G., et al., Efficacy and safety of the CVnCoV SARS-CoV-2 mRNA vaccine candidate in ten countries in Europe and Latin America (HERALD): a randomised, observer-blinded, placebo-controlled, phase 2b/3 trial. Lancet Infect Dis, 2022. 22(3): p. 329-340.
83. Cromer, D., et al., Relating In Vitro Neutralization Level and Protection in the CVnCoV (CUREVAC) Trial. Clin Infect Dis, 2022. 75(1): p. e878-e879.
84. Qu, L., et al., Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell, 2022. 185(10): p. 1728-1744 e16.
85. Erasmus, J.H., et al., An Alphavirus-derived replicon RNA vaccine induces SARS-CoV-2 neutralizing antibody and T cell responses in mice and nonhuman primates. Sci Transl Med, 2020. 12(555).
86. Rojas, L.A., et al., Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature, 2023. 618(7963): p. 144-150.
87. Sahin, U., et al., Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature, 2017. 547(7662): p. 222-226.
88. Sahin, U., et al., An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature, 2020. 585(7823): p. 107-112.
89. Holtkamp, S., et al., Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood, 2006. 108(13): p. 4009-17.
90. Barbier, A.J., et al., The clinical progress of mRNA vaccines and immunotherapies. Nat Biotechnol, 2022. 40(6): p. 840-854.
91. Liu, C., et al., mRNA-based cancer therapeutics. Nat Rev Cancer, 2023. 23(8): p. 526-543.
92. Lorentzen, C.L., et al., Clinical advances and ongoing trials on mRNA vaccines for cancer treatment. Lancet Oncol, 2022. 23(10): p. e450-e458.
The Nobel Assembly, consisting of 50 professors at Karolinska Institutet, awards the Nobel Prize in Physiology or Medicine. Its Nobel Committee evaluates the nominations. Since 1901 the Nobel Prize has been awarded to scientists who have made the most important discoveries for the benefit of humankind.
Nobel Prize® is the registered trademark of the Nobel Foundation
Nobel Prizes and laureates
Six prizes were awarded for achievements that have conferred the greatest benefit to humankind. The 14 laureates' work and discoveries range from quantum tunnelling to promoting democratic rights.
See them all presented here.

