Popular information
English
Swedish
![]()
The Nobel Prize in Chemistry 2004
A human cell contains some hundred thousand different proteins. These have numerous important functions: as accelerators of chemical reactions in the form of enzymes, as signal substances in the form of hormones, as important actors in the immune defence and by being responsible for the cell’s form and structure. This year’s Nobel Laureates in chemistry, Aaron Ciechanover, Avram Hershko and Irwin Rose, have contributed ground-breaking chemical knowledge of how the cell can regulate the presence of a certain protein by marking unwanted proteins with a label consisting of the polypeptide ubiquitin. Proteins so labelled are then broken down – degraded – rapidly in cellular “waste disposers” called proteasomes.
Through their discovery of this protein-regulating system Aaron Ciechanover, Avram Hershko and Irwin Rose have made it possible to understand at molecular level how the cell controls a number of very important biochemical processes such as the cell cycle, DNA repair, gene transcription and quality control of newly-produced proteins. New knowledge of this form of controlled protein death has also contributed to explaining how the immune defence functions. Defects in the system can lead to various diseases including some types of cancer.
Proteins labelled for destruction
Degradation needs no energy – or does it?
While great attention and much research have been spent on understanding how the cell controls the synthesis of a certain protein – at least five Nobel Prizes have been awarded in this area – the reverse, the degradation of proteins, has long been considered less important. A number of simple protein-degrading enzymes were already known. One example is trypsin, which in the small intestine breaks down proteins in our food to amino acids. Likewise, a type of cell organelle, the lysosome, in which proteins absorbed from outside are broken down, had long been studied. Common to these processes is that they do not require energy in order to function.
Experiments as long ago as the 1950s showed, however, that the breakdown of the cell’s own proteins does require energy. This long puzzled researchers, and it is precisely this paradox that underlies this year’s Nobel Prize in Chemistry: that the breakdown of proteins within the cell requires energy while other protein degradation takes place without added energy. A first step towards an explanation of this energy-dependent protein degradation was taken by Goldberg and his co-workers who in 1977 produced a cell-free extract from immature red blood cells, reticulocytes, which catalyse the breakdown of abnormal proteins in an ATP-dependent manner (ATP = adenosine triphosphate – the cell’s energy currency).
Using such an extract Aaron Ciechanover, Avram Hershko and Irwin Rose, in a series of epoch-making biochemical studies in the late 1970s and early 1980s, succeeded in showing that protein degradation in cells takes place in a series of step-wise reactions that result in the proteins to be destroyed being labelled with the polypeptide ubiquitin. This process enables the cell to break down unwanted proteins with high specificity, and it is this regulation that requires energy. As distinct from reversible protein modifications such as phosphorylation (Nobel Prize in Physiology or Medicine 1992), regulation through polyubiquitination is often irreversible since the target protein is destroyed. Much of the work was done during a series of sabbatical leaves that Avram Hershko and Aaron Ciechanover of the Technion (Israel Institute of Technology) spent with Irwin Rose at the Fox Chase Cancer Center in Philadelphia, USA.
The label is ubiquitin
The molecule that would later prove to be the label that marks out a protein for degradation was isolated as early as 1975. This 76-amino-acid-long polypeptide was isolated from calf sweetbread and was assumed to participate in the maturation of white blood cells. Since the molecule was subsequently found in numerous different tissues and organisms – but not in bacteria – it was given the name ubiquitin (from Latin ubique, “everywhere”) (fig. 1).

Fig 1. Ubiquitin – a common polypeptide that represents the “kiss of death”.
The discovery of ubiquitin-mediated protein degradation
After taking his doctorate, Avram Hershko had studied energy-dependent protein degradation in liver cells, but decided in 1977 to transfer to the reticulocyte extract described above. This extract contained large quantities of haemoglobin, which upset the experiments. In their attempts to remove the haemoglobin using chromatography, Aaron Ciechanover and Avram Hershko discovered that the extract could be divided into two fractions, each inactive on its own. But it turned out that as soon as the two fractions were recombined, the ATP-dependent protein degradation restarted. In 1978 the researchers reported that the active component of one fraction was a heat-stable polypeptide with a molecular weight of only 9000 which they termed APF-1 (active principle in fraction 1). This protein later proved to be ubiquitin.
The decisive breakthrough in the research was reported in two works that Ciechanover, Hershko and Rose published in 1980. Until that time the function of APF-1 was entirely unknown. In the first work it was shown that APF-1 was bound covalently, i.e. with a very stable chemical bond, to various proteins in the extract.
In the second work it was further shown that many APF-1 molecules could be bound to the same target protein; the latter phenomenon was termed polyubiquitination. We now know that this polyubiquitination of substrate proteins is the triggering signal that leads to degradation of the protein in the proteasome. It is this reaction that constitutes the actual labelling, the “kiss of death” if you will.
At a stroke, these entirely unanticipated discoveries changed the conditions for future work: it now became possible to concentrate on identifying the enzyme system that binds ubiquitin to its target proteins. Since ubiquitin occurs so generally in various tissues and organisms, it was quickly realised that ubiquitin-mediated protein degradation must be of general significance for the cell. In addition, the researchers guessed that the energy requirement in the form of ATP enabled the cell to control the specificity of the process.
The field was now open and between 1981 and 1983 Ciechanover, Hershko, Rose and their post docs and students developed “the multistep ubiquitin-tagging hypothesis” based on three newly-discovered enzyme activities they termed E1, E2 and E3 (fig. 2). We now know that a typical mammalian cell contains one or a few different E1 enzymes, some tens of E2 enzymes and several hundred different E3 enzymes. It is the specificity of the E3 enzyme that determines which proteins in the cell are to be marked for destruction in the proteasomes.
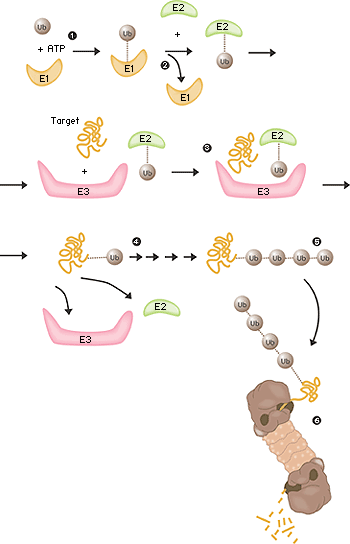
Fig 2. Ubiquitin-mediated protein degradation 1. The E1 enzyme activates the ubiquitin molecule. This reaction requires energy in the form of ATP. 2. The ubiquitin molecule is transferred to a different enzyme, E2. 3. The E3 enzyme can recognise the protein target which is to be destroyed. The E2-ubiquitin complex binds so near to the protein target that the actual ubiquitin label can be transferred from E2 to the target. 4. The E3 enzyme now releases the ubiquitin-labelled protein. 5. This last step is repeated until the protein has a short chain of ubiquitin molecules attached to itself. 6. This ubiquitin chain is recognised in the opening of the proteasome. The ubiquitin label is disconnected and the protein is admitted and chopped into small pieces.
All the studies up to this point had been done in cell-free systems. To be able to study the physiological function of ubiquitin-mediated protein degradation as well, Avram Hershko and his co-workers developed an immunochemical method. By using antibodies to ubiquitin, ubiquitin-protein-conjugate could be isolated from cells where the cell proteins had been pulse-labelled with a radioactive amino acid not present in ubiquitin. The results showed that cells really break down faulty proteins using the ubiquitin system, and we now know that up to 30% of the newly-synthesised proteins in a cell are broken down via the proteasomes since they do not pass the cell’s rigorous quality control.
The proteasome – the cell’s waste disposer
What is a proteasome? A human cell contains about 30,000 proteasomes: these barrel-formed structures can break down practically all proteins to 7-9-amino-acid-long peptides. The active surface of the proteasome is within the barrel where it is shielded from the rest of the cell. The only way in to the active surface is via the “lock”, which recognises polyubiquitinated proteins, denatures them with ATP energy and admits them to the barrel for disassembly once the ubiquitin label has been removed. The peptides formed are released from the other end of the proteasome. Thus the proteasome itself cannot choose proteins; it is chiefly the E3 enzyme that does this by ubiquitin-labelling the right protein for breakdown (fig. 3).
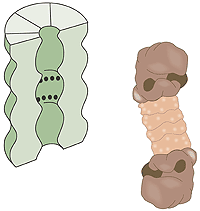
Fig 3. The cell’s waste disposer, the proteasome. The black spots indicate active, protein-degrading surfaces.
More recent research
While the biochemical mechanisms underlying ubiquitin-labelled protein degradation were laid bare around 1983 its physiological significance had not yet been fully understood. That it is of importance in destroying defective intracellular proteins was known but, to proceed, a mutated cell was needed in the ubiquitin system. By studying in detail how the mutated cell differs from a normal cell under various growth conditions, it was hoped to gain a better idea of what reactions in the cell depend on the ubiquitin system.
A mutated mouse cell had been isolated in 1980 by a research group in Tokyo. Their mouse-cell mutant contained a protein that, because of the mutation, was sensitive to temperature. At lower temperatures the protein functioned as it should, but not at higher. Cells cultured at the higher temperature stopped growing. In addition, they showed defective DNA synthesis and other erroneous functions at the higher temperature. Researchers in Boston quickly showed that the heat-sensitive protein in the mutant mouse cell was the ubiquitin-activating enzyme E1. Obviously, ubiquitin activation was necessary for the cell to function and reproduce itself at all. Controlled protein breakdown was not only important for degrading incorrect proteins in the cell but it probably also took part in control of the cell cycle, DNA replication and chromosome structure.
Since the late 1980s a number of physiologically important substrates for ubiquitin-mediated protein breakdown have been identified. Only a few of the most important will be mentioned here.
Prevention of self-pollination in plants
Most plants are bisexual, hermaphroditic. Self-pollination leads to a gradual decline in genetic diversity which in the long run can cause the whole species to die out. To prevent this, plants use ubiquitin-mediated degradation to reject “own” pollen. The exact mechanism has not yet been clarified but the E3 enzyme has been encountered and when proteasome inhibitors have been introduced, the rejection has been impaired.
Regulation of the cell cycle
When a cell is to make a copy of itself, many chemical reactions are involved. In a human being, six thousand million base pairs must be duplicated in DNA. These are gathered in 23 chromosome pairs that must be copied. Ordinary cell division, mitosis, and the formation of sex cells, meiosis, have many points of contact with the subjects of this year’s Nobel Prize. The E3 enzyme responsible, a protein complex termed the “anaphase-promoting complex” (APC) checks that the cell goes out of mitosis. This enzyme complex has also proved to play an important role in the separation of the chromosomes during mitosis and meiosis. A different protein complex acts like a rope around the chromosome pair, holding it together. At a given signal, the APC labels an inhibitor of a certain protein-degrading enzyme, whereupon the inhibitor is carried to the proteasome and destroyed. The enzyme is released, is activated and cuts the rope around the chromosome pair. Once the rope is gone, the chromosome pair can be separated. Incorrect chromosome division during meiosis is the commonest cause of spontaneous miscarriage during pregnancy, and an extra chromosome 21 in humans leads to Down’s syndrome. Most malignant tumours have cells with changed numbers of chromosomes as a result of incorrect chromosome division during mitosis.
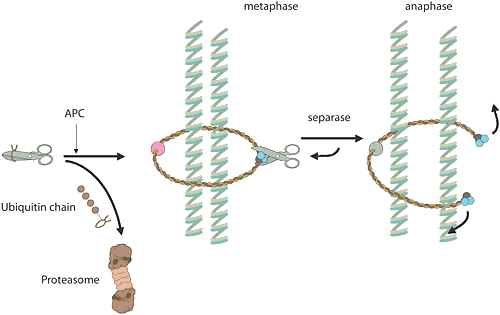
High resolution image (jpeg 230 kB)
DNA repair, cancer and programmed cell death
Protein p53 has been dubbed “the guardian of the genome” and it is a tumour-suppressor gene. This means that as long as a cell can produce p53 the development of cancer is hampered. Sure enough, the protein is mutated in at least 50% of all human cancer. The amount of protein p53 in a normal cell is low in consequence of continual production and breakdown. The breakdown is regulated through ubiquitination and the E3 enzyme responsible forms a complex with protein p53. Following DNA injury, protein p53 is phosphorylated and can no longer bind to its E3 enzyme. The breakdown stops and the quantity of p53 in the cell rises rapidly. Protein p53 acts as a transcription factor, i.e. a protein that controls the expression of a certain gene. Protein p53 binds to and controls genes that regulate DNA repair and programmed cell death. Raised levels of protein p53 lead first to interruption of the cell cycle to allow time for repair of DNA damage. If the damage is too extensive the cell triggers programmed cell death and “commits suicide”.
Infection with human papilloma virus correlates strongly to the occurrence of cervical cancer. The virus avoids the protein p53 control function through one of its proteins activating and changing the recognition pattern of a certain cellular E3 enzyme, E6-AP, which is tricked into ubiquitinating the protein p53, which is totally destroyed. In consequence of this the infected cell can no longer repair DNA damage in a normal manner or trigger programmed cell death. The DNA mutations increase in number and this can ultimately lead to the development of cancer.
Immune and inflammatory reactions
A certain transcription factor regulates many of the genes in the cell that are important for immune defence and inflammatory reactions. This protein, the transcription factor, occurs bound to an inhibitor protein in the cytoplasm of the cell, and the bound form of the transcription factor lacks activity. When cells are exposed to bacteria or various signal substances, the inhibitor protein is phosphorylated, and this results in its being ubiquitinated and broken down in the proteasome. The released transcription factor is transported to the cell nucleus where it binds to, and activates the expression of, specific genes.
The ubiquitin-proteasome system also produces the peptides that are presented by the immune defence on the surface of a virus-infected cell by breaking down virus proteins to suitable sizes. T lymphocytes recognise these peptides and attack the cell as an important part of our defence against virus infections.
Cystic fibrosis (CF)
The hereditary disease cystic fibrosis, CF, is caused by a non-functioning plasma membrane chloride channel called CFTR, the “cystic fibrosis transmembrane conductance regulator”. Most CF patients have one and the same genetic damage, loss of the amino acid phenylalanine in the CFTR protein. The mutation causes faulty folding of the protein and this in turn leads to the protein being retained in the cell’s control system for protein quality. This system ensures that the incorrectly folded protein is destroyed through ubiquitin-mediated protein breakdown instead of being transported out to the cell wall. A cell with no functioning chloride channel can no longer transport chloride ions through its wall. This affects secretion in, among other organs, the lungs and leads to the accretion of thick phlegm in the lungs which impairs their function, greatly increasing the risk of infection.
The ubiquitin system has become an interesting area of research for medicines against various diseases. Such preparations can be aimed at components of the ubiquitin-mediated breakdown system to prevent the degradation of specific proteins. They can also be designed to cause the system to destroy unwanted proteins. A medicine already being tested clinically is the proteasome inhibitor Velcade (PS341) which is used against multiple myeloma, a cancer disease that affects the body’s antigen-producing cells.
This year’s Laureates have explained the molecular background to a protein regulation system of great importance for all higher cells. New cell functions controlled by ubiquitin-mediated protein degradation are being discovered all the time and this research is being conducted in numerous laboratories all over the world.
| The Laureates | |
| Aaron Ciechanover | |
| Technion (Israel Institute of Technology) Rappaport Institute 1 Efron Street P.O. Box 9697 Haifa 31096 Israel |
Israeli citizen. Born 1947 (57 years) in Haifa, Israel. Doctor’s degree in medicine in 1975 at Hebrew University of Jerusalem, and in biology in 1982 at the Technion (Israel Institute of Technology), Haifa. Distinguished Professor at the Center for Cancer and Vascular Biology, the Rappaport Faculty of Medicine and Research Institute at the Technion, Haifa, Israel. |
| Avram Hershko | |
| Technion (Israel Institute of Technology) Rappaport Institute 1 Efron Street P.O. Box 9697 Haifa 31096 Israel |
Israeli citizen. Born 1937 (67 years) in Karcag, Hungary. Doctor’s degree in medicine in 1969 at the Hadassah and the Hebrew University Medical School, Jerusalem. Distinguished Professor at the Rappaport Family Institute for Research in Medical Sciences at the Technion, Haifa, Israel. |
| Irwin Rose | |
| Dept. of Physiology and Biophysics College of Medicine University of California, Irvine Irvine, CA 92697 USA |
American citizen. Born 1926 (78 years) in New York, USA. Doctor’s degree in in 1952 at the University of Chicago, USA. Specialist at the Department of Physiology and Biophysics, College of Medicine, University of California, Irvine, USA. |
Illustrations: Typoform
Nobel Prizes and laureates
Six prizes were awarded for achievements that have conferred the greatest benefit to humankind. The 14 laureates' work and discoveries range from quantum tunnelling to promoting democratic rights.
See them all presented here.

